GWAS 分析模型 | FaST-LMM
FaST-LMM (Factored Spectrally Transformed Linear Mixed Models) 是一个用于进行全基因组关联分析(GWAS)的模型。与标准混合线性模型相比,FaST-LMM 通过对遗传相似性矩阵进行单次谱分解来减少计算资源消耗并提升运行速度,因此特别适用于超大型数据集的 GWAS 分析。
扫码关注微信公众号【生信F3】获取更多生物信息学最新知识。

安装
FaST-LMM 提供了供直接运行的预编译版本(FastLmmC v2.07.20140723),下载后即可使用:
https://www.microsoft.com/en-us/download/details.aspx?id=52588
./fastlmmc
用法
FaST-LMM 需要四个输入文件(ASCII 编码),包含
-
SNP 数据 -
用于计算个体间遗传相似性(亲缘关系矩阵)的 SNP 数据(可以与1不同) -
表型数据 -
一组协变量(可选)
仅计算遗传相似性矩阵:
./fastlmmc -runGwasType NORUN \
-pheno Trait1.plink.txt -missingPhenotype NA \
-fileSim test \
-simOut test.sim
-runGwasType
计算遗传相似矩阵的谱分解后,运行 GWAS 或退出。使用 NORUN,缓存谱分解。默认值:RUN。
运行 GWAS:
./fastlmmc
-tfile test
-pheno Trait1.plink.txt
-tfileSim test
-simOut out.sim
-out test_fastlmm.out.txt
-missingPhenotype NA -maxChromosomeValue 1000000 -maxThreads 5
常用选项
1. 基因型
输入文件 SNP 数据应该为 PLINK 格式(ped/map, tped/tfam, bed/bim/fam, or fam/dat/map)。使用依赖 SNP 排序的二进制格式可以获得最快的速度。这些文件中的表型条目必须被设置为虚值并将被忽略(FaST-LMM 软件使用单独的表型文件)。性别应该被编码为一个数字。缺失的 SNP 值将被按均值填补。
注意:该软件只能接受整数或字符串形式(X、Y、XY 或 MT)的染色体编号,因此对于非模式物种,建议在 SNP 标识符中包含染色体 ID,并将染色体 ID 列全部以数字 0 填充。
-file basefilename
.map 和 .ped 格式的文件基名
-bfile basefilename
二进制 .bed, .fam 和 .bin 格式的文件基名
-tfile basefilename
转置后 .tfam 和 .tped 格式的文件基名
2. 亲缘关系
-tfileSim basefilename
用于构建遗传相似度(亲缘关系)的转置后 .tfam 和 .tped 格式的文件基名(可与基因型数据保持一致)
-simOut filename
指定将遗传相似度写入此文件
3. 表型
-pheno filename 表型文件名
-missingPhenotype
缺失值的标识符。如果一个个体的表型缺失,那么该个体就会被忽略。如果一个个体的协变量值缺失,则以平均值估算。默认值为 -9。
包含表型数据的文件使用 PLINK 的表型格式。其至少包含三列:familyID、individualID 和任意数量的表型值。列间以制表符或空格分隔。默认仅测试第一列表型值,缺失值默认以 -9 表示,但一般建议以 -missingPhenotype 选项来指定缺失值。第一列 familyID 与第二列 individualID 相连接从而为个体创建唯一的标识符,并与上述 PLINK 文件中的个体条目相匹配。例如:
cid0P0 cid0P0 0.4853395139922632
cid1P0 cid1P0 -0.2076984565752155
cid2P0 cid2P0 1.4909084058931985
cid3P0 cid3P0 -1.2128996652683697
cid4P0 cid4P0 0.4293203431508744
4. 其它
-maxThreads <int>
该选项被传递给 MKL 数学库(Intel)以 "建议" 使用的并行程度。指定一个大于计算机上核心数的数字可能会导致程序运行得更慢。指定一个小于核心数的数字可能会使计算机在运行 FastLmmC 时不会在程序的不同阶段消耗所有的 CPU 资源。在使用 ACML 数学库(AMD)时,MaxThreads 选项将被忽略。
-covar filename
包含协变量的可选文件
结果输出
-out filename
输出文件的名称。默认值是 [basefilename].out.txt。如果使用扩展名是 .csv,输出文件将以逗号分隔。否则将以制表符分隔。
默认输出文件如下所示:
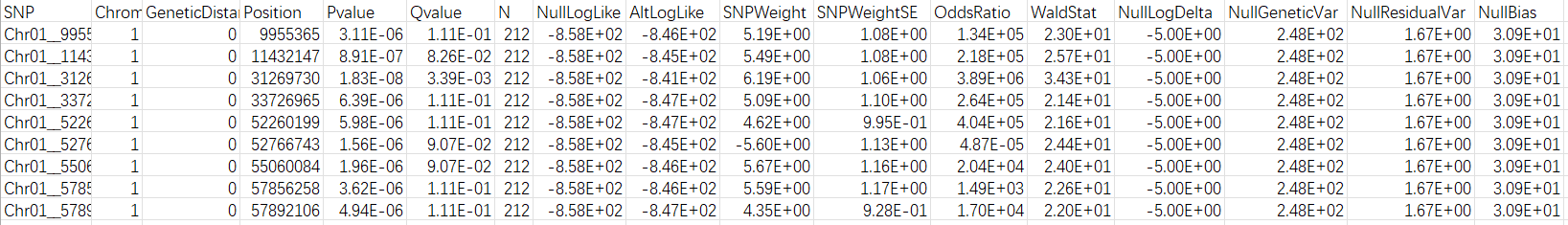
每列含义如下:
-
SNP:SNP 标识符。 -
Chromosome:SNP 的染色体标识符,取自 PLINK 文件。 -
Genetic Distance:SNP 的遗传距离,未知则为 0。 -
Position:SNP 的物理位置。 -
Pvalue:计算所得 P 值。 -
Qvalue:使用 Benjamini-Hochberg 校正 p 值后所得的 q 值 -
N:用于分析的个体数量 -
NullLogLike -
AltLogLike -
SNPWeight -
SNPWeightSE -
OddsRatio -
WaldStat -
NullLogDelta -
NullGeneticVar -
NullResidualVar -
NullBias
扫码关注微信公众号【生信F3】获取更多生物信息学最新知识。
本文由 mdnice 多平台发布