文章目录
- Salmon简介
- 文章引用及适用物种
- 获取转录组并建立索引
- 比较salmon和coverm定量差异
Salmon简介
Salmon 是一款速度极快的程序,能从 RNA-seq 数据中生成高精度的转录本水平的量化估计值。Salmon 通过一系列不同的创新实现了其准确性和速度,包括使用选择性比对(准确的传统读段比对的但快速计算的替代方法)和大规模并行随机坍缩变异推断。因此,该工具用途广泛,能很好地融入许多不同的工作流程。例如,您可以选择使用我们的选择性比对算法,向 Salmon 提供原始测序读数,或者,如果更方便的话,您可以向 Salmon 提供常规比对(例如,用您最喜欢的比对器生成的与转录组比对的未排序 BAM 文件),Salmon 将使用同样快速、最先进的推断算法为您的实验估算转录本水平丰度。
- salmon也可对核酸的contig序列索引,6G的参考基因组,双端60G fq文件耗时仅仅16m(150 core)

文章引用及适用物种
Salmon于2017年在Nature Methods发表以来,Salmon provides fast and bias-aware quantification of transcript expression, Nature Methods, 2017已经被多次引用,其中包括在人、细菌及真菌等多个物种。
获取转录组并建立索引
为了量化转录本水平的丰度,Salmon需要目标转录组。 该转录组以多FASTA序列文件(可以是压缩的)的形式提供给Salmon,每个序列均是转录本。 在此示例中,我们将分析拟南芥的一些数据,因此我们将下载拟南芥转录组并为其编制索引。
- 下载参考转录组
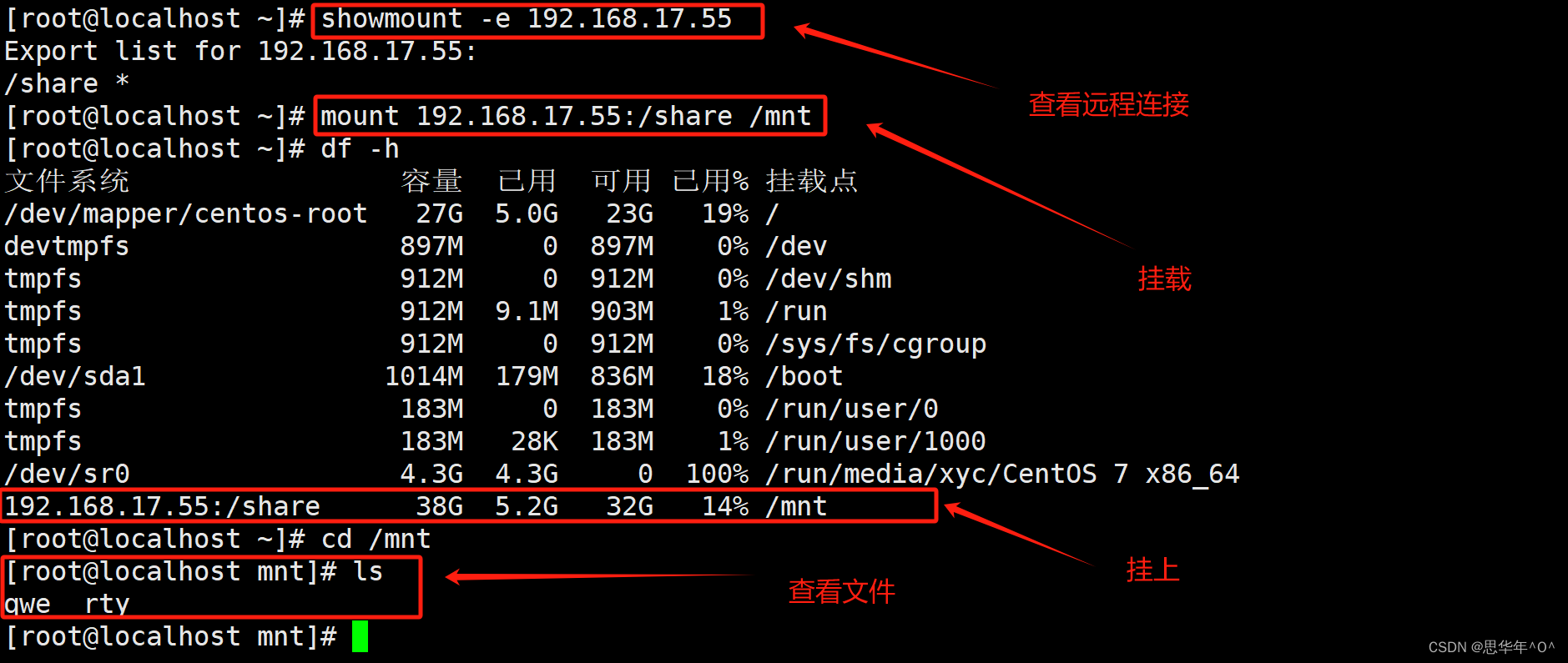
curl ftp://ftp.ensemblgenomes.org/pub/plants/release-28/fasta/arabidopsis_thaliana/cdna/Arabidopsis_thaliana.TAIR10.28.cdna.all.fa.gz -o athal.fa.gz
- 构建index
Next, we’re going to build an index on our transcriptome. The index is a structure that salmon uses to quasi-map RNA-seq reads during quantification. The index need only be constructed once per transcriptome, and it can then be reused to quantify many experiments. We use the index command of salmon to build our index:
salmon index -p 40 -t athal.fa.gz -i athal_index
-t [ --transcripts ] arg Transcript fasta file.
-i [ --index ] arg salmon index.
-p [ --threads ] arg (=2) Number of threads to use during indexing.
耗时:1m35.946s
生成athal_index目录:

- 注意:若构建index时不加
--keepDuplicates,则会去掉一些重复的转录本,导致最后定量的转录本数量比输入的少,会在log日志记录这些信息
see here
[2021-07-22 17:46:23.310] [jointLog] [warning] Removed 351 transcripts that were sequence duplicates of indexed transcripts.
[2021-07-22 17:46:23.310] [jointLog] [warning] If you wish to retain duplicate transcripts, please use the `--keepDuplicates` flag
去重复的转录本保存在duplicate_cluster.tsv,第一列是保存的转录本,第二列是去掉的重复片段,以下是一个例子,保留的是RWS.20.06_k141_357096_5,发现两条序列确实一样,可能是因为之前去冗余残留了极少量这类片段
>RWS.20.06_k141_357096_5 # 2137 # 2244 # 1 # ID=776723_5;partial=00;start_type=ATG;rbs_motif=AGGAG;rbs_spacer=5-10bp;gc_cont=0.630
MPEARQEHTTQRMSRVDTAWLRMDNDVNLMMIVGV*
>TGLI3.17_k141_676506_1 # 66 # 173 # -1 # ID=111959_1;partial=00;start_type=ATG;rbs_motif=AGGAG;rbs_spacer=5-10bp;gc_cont=0.639
MPEARQEHTTQRMSRVDTAWLRMDNDVNLMMIVGV*
- 定量paired sequence
salmon quant -i athal_index -l A -1 ${samp}_1.fastq.gz -2 ${samp}_2.fastq.gz -p 8 --validateMappings -o ${samp}_quant
参数解释:
-i: salmon index
-l A: 告诉Salmon,它应该自动确定测序reads的库类型(例如,链状与非链状等)。(e.g. stranded vs. unstranded etc.).
-1 and -2: 参数告诉salmon在哪里可以找到这个样本的左右读数(注意,salmon会直接接受gzipped FASTQ文件)
-p 8: threads
-o: 输出目录
--validateMappings: [Quasi-mapping mode only] ,使用基于比对的核实。如果这个传递这个标志,标准映射将被验证以确保它们能产生一个合理的比对方式,然后再进一步用于量化
结果:
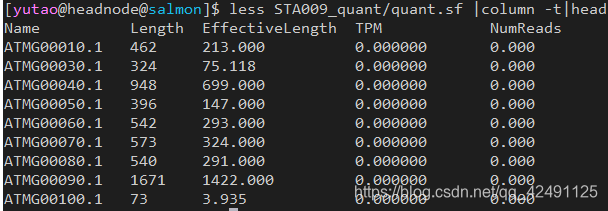
主要的输出文件(称为quant.sf)是相当不言自明的。例如,看一下quants/DRR016125/quant.sf中DRR016125样本的定量文件,你会看到一个简单的TSV格式文件,列出每个转录本的名称(Name)、长度(Length)、有效长度(EffectiveLength)(更多细节见文档),以及它在每百万转录本(TPM)中的丰度和源自该转录本的估计读数(NumReads)。
比较salmon和coverm定量差异
(salmon) [yutao@globin test]$ ll ../GGG_1134OTUs_prokka/ERR1201173_maxbin2_bin.0.ffn
-rw-r--r-- 1 yutao husn 5.6M Feb 28 23:44 ../GGG_1134OTUs_prokka/ERR1201173_maxbin2_bin.0.ffn
(salmon) [yutao@globin test]$ ll ../GGG_130samples_10k_fastq/ERR1201173_?.fastq
-rw-r--r-- 1 yutao husn 2.5M Feb 7 20:12 ../GGG_130samples_10k_fastq/ERR1201173_1.fastq
-rw-r--r-- 1 yutao husn 2.5M Feb 7 20:13 ../GGG_130samples_10k_fastq/ERR1201173_2.fastq
# coverm
(coverm) [yutao@globin test]$ time coverm contig -1 ../GGG_130samples_10k_fastq/ERR1201173_1.fastq -2 ../GGG_130samples_10k_fastq/ERR1201173_2.fastq --reference ERR1201173_maxbin2_bin.0.ffn --methods count --output-file ERR1201173_coverm_tpm.tsv --no-zeros
[2023-03-03T07:23:32Z INFO coverm] CoverM version 0.6.1
[2023-03-03T07:23:32Z INFO coverm] Writing output to file: ERR1201173_coverm_tpm.tsv
[2023-03-03T07:23:32Z INFO coverm] Using min-covered-fraction 0%
[2023-03-03T07:23:32Z INFO bird_tool_utils::external_command_checker] Found minimap2 version 2.17-r941
[2023-03-03T07:23:32Z INFO bird_tool_utils::external_command_checker] Found samtools version 1.14
[2023-03-03T07:23:32Z INFO coverm] Not pre-generating minimap2 index
[2023-03-03T07:23:33Z INFO coverm::contig] In sample 'ERR1201173_maxbin2_bin.0.ffn/ERR1201173_1.fastq', found 3904 reads mapped out of 20000 total (19.52%)
# salmon
(salmon) [yutao@globin test]$ time salmon quant -i ERR1201173_maxbin2_bin.0_salmon/ -l A -1 ../GGG_130samples_10k_fastq/ERR1201173_1.fastq -2 ../GGG_130samples_10k_fastq/ERR1201173_2.fastq -p 8 --validateMappings -o ERR1201173_salmon
(salmon) [yutao@globin test]$ time salmon quant -i ERR1201173_maxbin2_bin.0_salmon/ -l A -1 ../GGG_130samples_10k_fastq/ERR1201173_1.fastq -2 ../GGG_130samples_10k_fastq/ERR1201173_2.fastq -p 8 --validateMappings -o ERR1201173_salmon
# Mapping rate = 18.9%
# 比较(基于reads count)
# 1.coverm比salmon定量到更多序列(covem可能更灵敏)
(coverm) [yutao@globin test]$ awk '$5>0' ERR1201173_salmon/quant.sf |wc -l
1425
(coverm) [yutao@globin test]$ awk '$2>0' ERR1201173_coverm_tpm.tsv|wc -l
1905
# 每个序列平均丰度coverm更高
(coverm) [yutao@globin test]$ awk '{sum+=$2}END{print sum/NR}' ERR1201173_coverm_tpm.tsv
2.04934
(coverm) [yutao@globin test]$ awk '{sum+=$5}END{print sum/NR}' ERR1201173_salmon/quant.sf
0.303322
# 2.定量最大的序列二者基本一致
(coverm) [yutao@globin test]$ sort -k2 -nr ERR1201173_coverm_tpm.tsv|head
CMLNLAJI_01761 36
CMLNLAJI_03398 23
CMLNLAJI_00817 17
CMLNLAJI_03401 16
CMLNLAJI_03403 11
CMLNLAJI_05922 10
CMLNLAJI_05660 10
CMLNLAJI_00085 10
CMLNLAJI_03402 9
CMLNLAJI_03067 9
(coverm) [yutao@globin test]$ sort -k5 -nr ERR1201173_salmon/quant.sf |head
CMLNLAJI_01761 924 526.586 4518.043434 18.000
CMLNLAJI_03398 1542 1144.360 1386.006957 12.000
CMLNLAJI_00817 396 65.543 24199.067240 12.000
CMLNLAJI_03401 1284 886.360 1192.962147 8.000
CMLNLAJI_03403 609 216.428 4274.954676 7.000
CMLNLAJI_03402 528 146.759 5403.730684 6.000
CMLNLAJI_05922 1500 1102.360 599.505802 5.000
CMLNLAJI_05660 2899 2501.360 264.204769 5.000
CMLNLAJI_03067 1428 1030.360 641.398359 5.000
CMLNLAJI_00085 4419 4021.360 164.340235 5.000
# tpm比较:salmon定量的更大(大概大10倍)
(coverm) [yutao@globin test]$ sort -k4 -nr ERR1201173_salmon/quant.sf |head
CMLNLAJI_00817 396 65.543 24199.067240 12.000
CMLNLAJI_04753 117 10.074 13120.238381 1.000
CMLNLAJI_03636 120 13.074 10109.645579 1.000
CMLNLAJI_03582 336 45.517 8711.538955 3.000
CMLNLAJI_01682 198 32.678 8089.537307 2.000
CMLNLAJI_06231 258 38.674 6835.329307 2.000
CMLNLAJI_01988 273 40.469 6532.134741 2.000
CMLNLAJI_02618 231 42.394 6235.487025 2.000
CMLNLAJI_03253 291 43.659 6054.827349 2.000
CMLNLAJI_03808 297 45.390 5823.973634 2.000
(coverm) [yutao@globin test]$ sort -k2 -nr ERR1201173_coverm_tpm.tsv|head
CMLNLAJI_00817 9356.189
CMLNLAJI_01761 8491.334
CMLNLAJI_00818 6918.8647
CMLNLAJI_00737 6054.0054
CMLNLAJI_03403 3936.5942
CMLNLAJI_03402 3714.958
CMLNLAJI_03398 3250.789
CMLNLAJI_04876 3228.8027
CMLNLAJI_05910 2985.5366
CMLNLAJI_05661 2945.1917
参考
Salmon provides fast and bias-aware quantification of transcript expression, Nature Methods, 2017
Salmon github
Salmon tutorial