基因组组装完成后,或者是完成了草图,就不可避免遇到一个问题,需要对基因组序列进行注释。注释之前首先得构建基因模型,有三种策略:
从头注释(de novo prediction):通过已有的概率模型来预测基因结构,在预测剪切位点和UTR区准确性较低
同源预测(homology-based prediction):有一些基因蛋白在相近物种间的保守型搞,所以可以使用已有的高质量近缘物种注释信息通过序列联配的方式确定外显子边界和剪切位点
基于转录组预测(transcriptome-based prediction):通过物种的RNA-seq数据辅助注释,能够较为准确的确定剪切位点和外显子区域。
每一种方法都有自己的优缺点,所以最后需要用EvidenceModeler(EVM)和GLEAN工具进行整合,合并成完整的基因结构。基于可靠的基因结构,后续可才是功能注释,蛋白功能域注释,基因本体论注释,通路注释等。
例如:Cardamine hirsuta(碎米荠)基因组注释
文章标题为“The Cardamine hirsuta genome offers insight into the evolution of morphological diversity”。
同源注释:使用 GenomeThreader 以拟南芥为剪切模型,以及PlantsGDB resourc上 Brassica rapa (v1.1), A. thaliana(TAIR10), A. lyrata (v6), tomato (v3.6), poplar (v2) 和 A. thaliana (version PUT-169), B. napus (version PUT-172) EST assemblies 的完整的代表性蛋白集。
转录本预测: 将 C. hirsuta RNA-seq数据比对到基因序列,然后用cufflinks拼接
从头预测:转录本预测得到的潜在蛋白编码转录本使用网页工具 ORFpredictor 进行预测, 同时用 blastx 和 A. thalina 进行比较,选择90%序列相似度和最高5%长度差异的部分从而保证保留完整的编码框(有启动子和终止子)。 这些基因模型根据相互之间的相似度和重叠度进行聚类,高度相似(>95)从聚类中剔除,保证非冗余训练集。为了训练gene finder, 它们选随机选取了2000个位点,20%是单个外显子基因。从头预测工具为 August , GlimmerHMM, Geneid 和 SNAP . 此外还用了Fgenesh+, 以双子叶特异矩阵为参数进行预测。
最后使用JIGSAW算法根据以上结果进行训练,随后再次用JIGSAW对每个基因模型计算统计学权重。
可变剪切模型则是基于苗、叶、花和果实的RNA-seq比对组装结果。
GO注释使用AHRD流程
参考 原文
定义
基因组注释:是利用生物信息学方法和工具,对基因组所有基因的生物学功能进行高通量注释,是当前功能基因组学研究的一个热点。
基因组注释:即在一条DNA序列上,通过从头、同源、结构定义等多种方法,搜寻并定义基因组原件,得到其位置、序列、结构、功能等信息。
基因组注释流程图
基因组注释前期准备
物种拉丁名,例如:Orazy sativa,基因id:Osa000001
同源物种: 一般选5个左右物种,需要有注释的基因/蛋白序列,保证高组装和注释质量
转库组数据: RNAseq和lsoseq注释(用于结构注释中的转录辅助注释)(建议自测同样本的数据)
基因组注释的分析内容
]
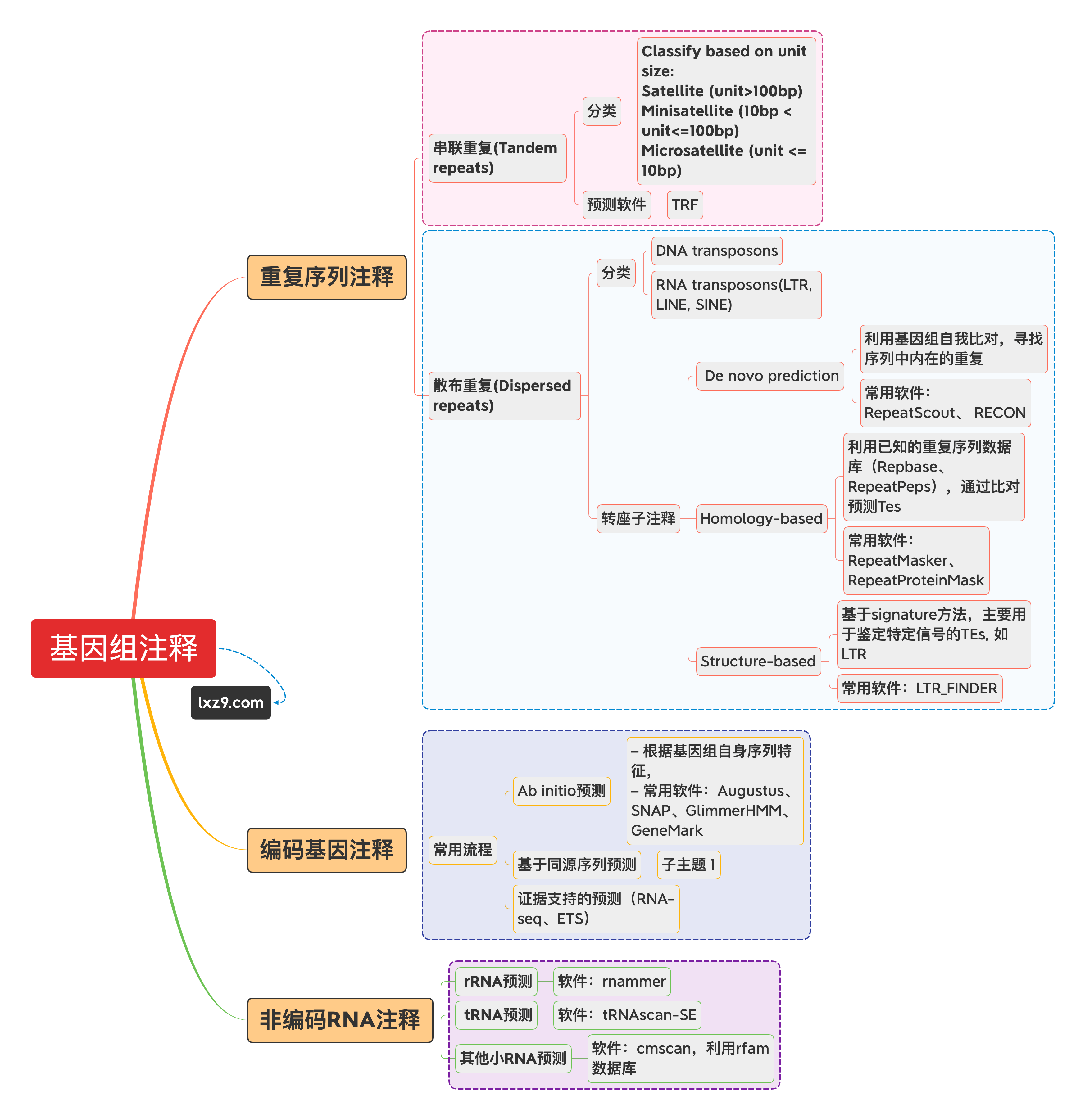
重复注释
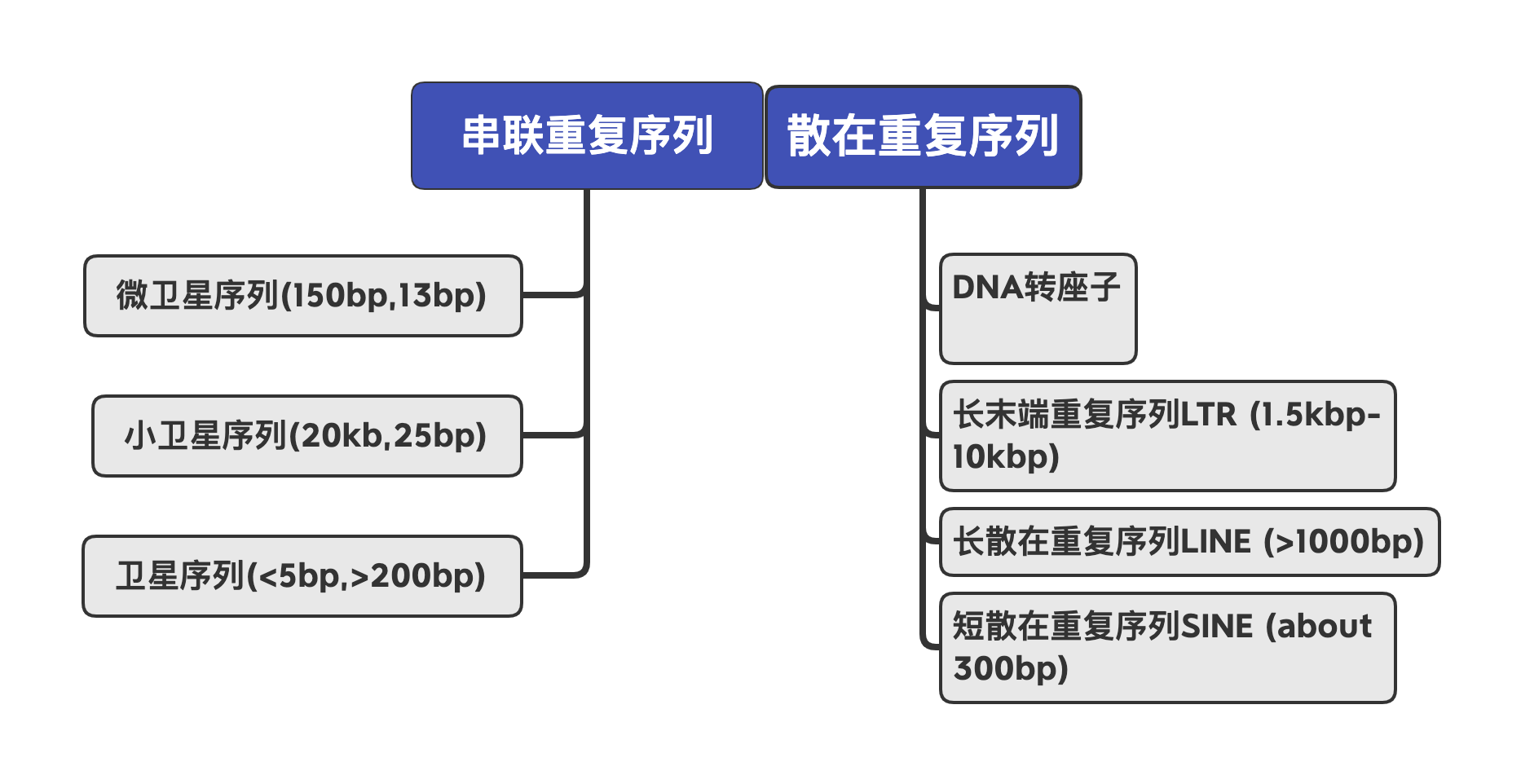
重复序列广泛存在于真核生物基因组中,这些重复序列或集中成簇,或分散在基因之间。根据分布把重复序列分为散在重复序列和串联重复序列。
重复序列根据序列特征分为2类:串联重复(Tandem repeats)和散布重复(Dispersed repeats)
- RepeatMasker:基于Repbase(dna)/自建elibrary查询重复序列
1 2 3 | RepeatMasker -nolow -no_is -norna -parallel 2 -lib RepeatMasker.lib genome.fa #-nohow:屏蔽低复杂简单重复; -no_is:跳过细菌插入元件检查; -norna:不掩盖小RNA(伪)基因; #-parallel 并行使用的处理器数,可提升分析速度 |
- RepeatProteinMask:基于 Repbase(pep)查询重复序列
1 2 3 | RepeatProteinMask -noLowSimple -pvalue 0.0001 genome.fa #noLowSimple:关闭低复杂度和简单重复的屏蔽/注释; -pvalue:接受匹配的阈值 #注意点: genome.fa的D不能长于18个字符 |
- TRF:元件的结构特征等来识别重复序列
1 | trf genome.fa 2 7 7 80 10 50 2000 -d -h |
- LTR-FINDER:基于重复序列特征
1 2 | Itr_finder -W 2 -C -s tRNAs.fa genome.fa
#-w 2 输出格式,2-table; -C:检测中心粒,删除高重复区域
|
- repeatmodeler:基于自身序列比对
1 2 3 4 | BuildDatabase -name mydb genome.fa RepeatModeler -database mydb -pa 6 >run.out #-name:创建 database的名称; #-pa:共享内存处理器的数量程序,可提升分析速度 |
| 每个软件都有很多参数,可-help/-h自行查看,参数的选择最好是参考已发表的文献 |
结构注释
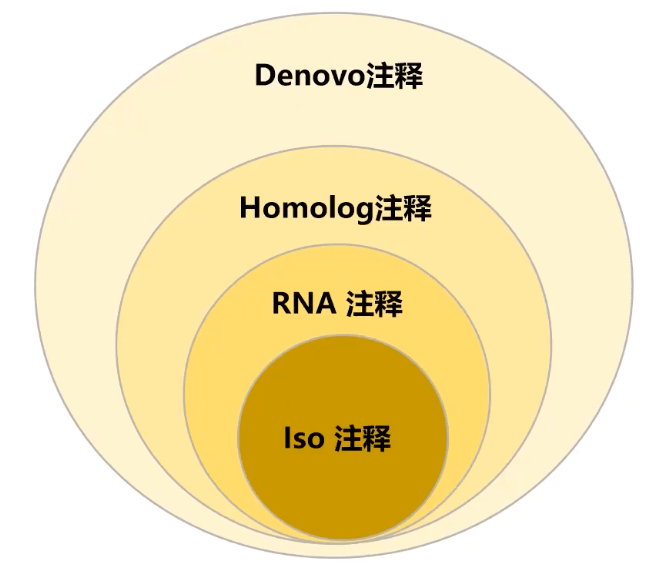
结构注释:注释可以产生具有生物学功能的蛋白的基因。一般包括启动子,转录起始,5’UTR,起始密码子,外显子,内含子,终止密码子,3’UTR,poly-A等结构。
]
De novo预测(屏蔽重复序列)
- Augustus(真核)
1 2 3 4 | augustus --species=XXX --AUGUSTUS CONFIG PATH= config --uniqueGeneld=true --nolnFrameStop=true--gff3=on --strand=both genome.mask.fa> genome.mask.fa.out # --uniqueGeneld=true:gene:命名 aseqname.gn; # --nolnFrameStop=true:不带有终止密码子的转录本; # --gff3=on:输出格式gff3 |
- GlimmerHMM(真核,预测的基因数目较多长度较短,一般用于植物)
1 2 3 4 5 6 | glimmerhmm.genome.mask.fa -d XXX- f -g genome.mask.fa.gff # -d 库de路径; # -f:不要partial gene predictions; # -g输出格式gff |
- Genscan(真核,其预测的内含子较大,一般用于动物)
1 2 | genscan Humanlso.smat genome.mask.fa > genome.mask.fa.genscan
# Humanlsc.smat:参数文件,软件自带
|
4.其他软件
SNAP. GenelD GenemarkS
denovo的软件很多,两个软件就可以了,太多软件会增加较多的假阳性,一般在
Augustus, GlimmerHMM, Genscan中选择即可
Homolog注释
利用近缘物种已知基因进行序列比对,找到同源序列。然后在同源序列的基础上,根据基因信号如剪切信号、基因起始和终止密码子对基因结构进行预测。
相对于从头预测的“大海捞针”,同源预测相当于先用一块磁铁在基因组大海中缩小了可能区域,然后从可能区域中鉴定基因结构。
利用TBlastn将同源物种的蛋白比对回基因组,得到候选区域。
利用 EXonerate/ Genewise进行精确的蛋白-核酸比对,以得到剪接位点。
Exonerate解决了 GeneWisez存在的很多问题,并且速度快了1000倍,默认选择EXonerate分析
RNA-seq辅助注释
tophat比对————>cufflink转录本————>TransDecoder
- 将RNAseq数据进行tophat比对;
- 比对后的结果文件利用cufflink构建转录本
- 使用TransDecoder在构建的转录本上预测Open Reading Frame(ORF)。
Iso seq 辅助注释
CD-HIT————>gmap比对————>TransDecoder
- 将物种的三代全长转录本用CD-HIT进行去冗余;
- 将去冗余后的序列使用gmap比对回基因组得到转录本位置;
- 使用TransDecoder在构建的转录本上预测 Open Reading Frame(ORF).
基因结构预测方法可信度排序
MAKERE整合
在基因组注释上, MAKER算是一个很强大的分析流程,主要是进行 Denovo注释, Homolog注释,转录辅助注释三者的整合,保证最终注释基因集的可靠性
1 2 3 4 | maker maker_exe.ctl maker_opts.ctl maker_bopts.ctl #maker exe.ct:执行程序的路径 #maker_ boots.ctl: BLAST7和 Exonerate的过滤参数 #maker opts.ctl:其他信息,例如输入基因组文件,主要调整输入文件等( genome= ;est= ;protein= ;pred_gff= ;) |
nCRNA注释
- rRNA(核糖体RNA)
与蛋白质结合形成核糖体,其功能是作为mn的支架,提供mRNA翻译成蛋白质的场所。 - tRNA(转运RNA)
·携带氨基酸进入核糖体,使之在mRNA指导下合成蛋白质。 - miRNA(miRNA)
·将mRNA降解或抑制其翻译,具有沉默基因的功能。 - SnRNA(小核RNA)
·主要参与RNA前体的加工过程,是RNA剪切体的主要成分。
miRNA与snRNA注释
- 采用Rfam和INFERNAL进行二级结构检测。
- ftp://ftp.sanger.ac.uk/pub/databases/Rfam
- blastn+cmsearch (INFERNAL程序)
rRNA注释
- 由于rRNA的结构保守程度非常高,因此采用与已有的全长rRNA进行blastn比对而获得。
- blastn
tRNA注释
- 结构特点:三叶草型二级结构。
- 预测方法:针对二级结构进行检测。使用tRNAscan-SE
功能注释
功能注释:基因功能的注释依赖于上一步的基因结构预测,根据预测结果从基因组上提取翻译后的蛋白序列和主流的数据库进行blastp比对,完成功能注释。
常用数据库一共有以下几种:NR,KEGG, Uniprot (Swiss-Prot, TrEMBL),InterPro,Go
- KEGG
- 生物学通路数据库(Gene,Pathway,Ligand).
- KEGG: Kyoto Encyclopedia of Genes and Genomes
- blastp
- SWISS-PROT和TrEMBL
- UniProt (Universal Protein Resource)蛋白质序列数据库PIR、SWISS-PROT和TrEMBL统一起来,建立了一个蛋白质数据库。
- UniProt
- blastp
- Interpro
- 蛋白家族(protein families)、功能保守区域(domains)和功能位点(funtional sites)的数据库.
- InterPro
- InterProScan
- GO
- 基因功能注释数据库(GeneOntology)
- 三个层面Cellular Component、 Biological Process、 Molecular Function.
- Gene Ontology Resource
- InterProScan
基因组评估
- BUSCO评估
BUSCO是一款使用python语言编写的对转录组和基因组组装质量进行评估的软件。在相近的物种之间总有一些保守的序列,而BUSCO就是使用这些保守序列与组装的结果进行比对,鉴定组装的结果是否包含这些序列,包含单条、多条还是部分或者不包含等等情况来给出结果。
BUSCO软件根据OrthoDB数据库,构建了几个大的进化分支的单拷贝基因集。将其与该基因集进行比较,根据比对上的比例、完整性,来评价准确性和完整性。
总结
基因组注释
重复注释————RepeatMask, RepeatProteinMask, TRF, LTR-FINDER,repeatmodeler;
结构注释————Denovo注释,同源注释,转录辅助注释;
ncRNA注释————tRNA,rRNA,miRNA,snRNA;
功能注释————NR, KEGG, InterPro,SWISS-PROT,TrEMBL,GO;
基因组评估————BUSCO
附录
基因组注释的常用软件:
重复区域:
RepeatMasker:识别基因组中的可能重复
RepeatModeler: 识别新的重复序列
LTR-FINDER: http://tlife.fudan.edu.cn/ltr_finder/
从头预测:
Augustus
Fgenesh
同源预测:
GeneWise
Exonerate
Trinity
GenomeThreader
注释合并:
GLEAN:已经落伍于时代了
EvidenceModeler: 与时俱进
流程
PASA:真核生物基因的转录本可变剪切自动化注释项目,需要提供物种的EST或RNA-seq数据
MAKER
BRAKER1: 使用GeneMark-ET和AUGUSTUS基于RNA-Seq注释基因结构
EuGene
可视化
IGV
JBrowse/GBrowse
参考文献和推荐阅读:
NCBI真核生物基因组注释流程https://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/
真核基因组注释入门: “A beginner’s guide to eukaryotic genome annotation”
二代测序注释流程:Comparative Gene Finding: “Annotation Pipelines for Next-Generation Sequencing Projects”
基因组转录组注释策略: “Plant genome and transcriptome annotations: from misconceptions to simple solution”
重复序列综述: “Repetitive DNA and next-generation sequencing: computational challenges and solutions”
MAKER2教程: http://weatherby.genetics.utah.edu/MAKER/wiki/index.php/MAKER_Tutorial_for_WGS_Assembly_and_Annotation_Winter_School_2018
《生物信息学》 樊龙江: 第1-5章: 基因预测与功能注释
《NGS生物信息分析》 陈连福: 真核生物基因组基因注释
JGS流程: https://genome.jgi.doe.gov/programs/fungi/FungalGenomeAnnotationSOP.pdf