Biopython教程

参考: https://biopython-cn.readthedocs.io/zh_CN/latest/index.html
蛋白质文件获取
- Entrez方法
from Bio import Entrez
Entrez.email='邮箱名' #如'123456789@qq.com'
handle=Entrez.esearch(db='protein',term='2rbg')
record=Entrez.read(handle)
id=record['IdList'][0]
handle=Entrez.efetch(db='protein',id=id,rettype='gb',retmode='text')
with open('2rbg.pdb','w') as f:
for i in handle.readlines():
f.write(i)
- 直接获取数据文件
retrieve_pdb_file
from Bio import PDB
PDB.PDBList().retrieve_pdb_file(pdb_code='2FAT',file_format='mmCif')
retrieve_pdb_file函数中pdb_code表示蛋白文件名,file_format表示文件格式,如’‘pdb’、'mmCif’等。如果需要下载到指定文件夹,需要使用参数pdir,参数值为指定文件夹的绝对路径。
io = PDBIO()
io.set_structure(structure)
io.save('out.pdb')
如果想输出结构的一部分,可以用 Select 类(也在 PDBIO 中)来实现。 Select 有如下四种方法:
- accept_model(model)
- accept_chain(chain)
- accept_residue(residue)
- accept_atom(atom)
在默认情况下,每种方法的返回值都为1(表示model/chain/residue/atom被包含在输出结果中)。通过子类化 Select 和返回值0,你可以从输出中排除model、chain等。也许麻烦,但很强大。接下来的代码将只输出甘氨酸残基:
class GlySelect(Select):
def accept_residue(self, residue):
if residue.get_name()=='GLY':
return True
else:
return False
io = PDBIO()
io.set_structure(structure)
io.save('gly_only.pdb', GlySelect())
如果这部分对你来说太复杂,那么 Dice 模块有一个很方便的 extract 函数,它可以输出一条链中起始和终止氨基酸残基之间的所有氨基酸残基。
蛋白质结构写PDB文件
可以用PDBIO类实现。当然也可很方便地输出一个结构的特定部分。
蛋白质结构解析
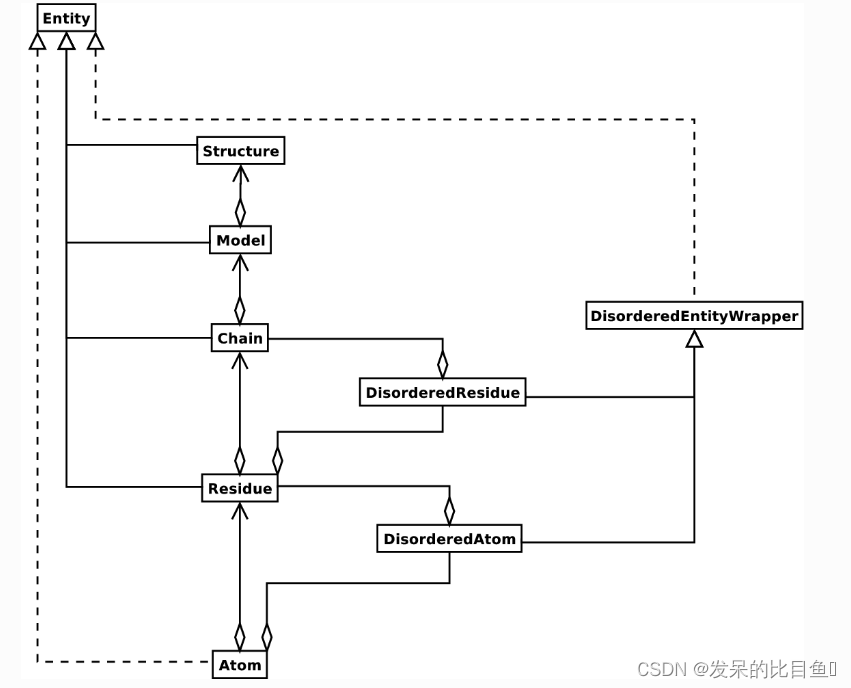
一个 Structure 对象的整体布局遵循称为SMCRA(Structure/Model/Chain/Residue/Atom,结构/模型/链/残基/原子)的体系架构:
- 结构由模型组成
- 模型由多条链组成
- 链由残基组成
- 多个原子构成残基
一个 Structure 对象的UML图(暂时忘掉 Disordered 吧)如上图所示。这样的数据结构不一定最适用于表示一个结构的生物大分子内容,但要很好地解释一个描述结构的文件中所呈现的数据(最典型的如PDB或MMCIF文件),这样的数据结构就是必要的了。如果这种层次结构不能表示一个结构文件的内容,那么可以相当确定是这个文件有错误或至少描述结构不够明确。一旦不能生成SMCRA数据结构,就有理由怀疑出了故障。因此,解析PDB文件可用于检测可能的故障。
结构,模型,链,残基都是实体基类的子类。原子类仅仅(部分)实现了实体接口(因为原子类没有子类)。
对于每个实体子类,你可以用该子类的一个唯一标识符作为键来提取子类(比如,可以用原子名称作为键从残基对象中提取一个原子对象;用链的标识符作为键从域对象中提取链)。
紊乱原子和残基用DisorderedAtom和DisorderedResidue类来表示,二者都是DisorderedEntityWrapper基类的子类。它们隐藏了紊乱的复杂性,表现得与原子和残基对象无二。
获取structure类对象
from Bio import PDB
parser=PDB.PDBParser(QUIET=True)
structure=parser.get_structure(file='2rbg.pdb',id=None)
先使用PDBParser()创建PDB解析器parser,再用parser从pdb文件中解析蛋白质结构,得到structure类对象。parser.get_structure()函数中file参数表示解析的蛋白质文件,id是一个编号,可以用数、字符串等标记,也可以用None。得到的structure用于后面肽链chain、氨基酸残基residues、原子atom、模型model等数据的获取。
注:除了pdb文件,对于其它类型的文件,可以使用相应的PDB.MMCIFParser()、PDB.FastMMCIFParser()解析器,使用get_structure()得到structure,之后的方法都是相同的。
许多PDB文件头包含不完整或错误的信息。许多错误在等价的mmCIF格式文件中得到修正。 因此,如果你对文件头信息感兴趣, MMCIF2Dict 来提取信息,而不用处理PDB文件文件头。
full_id 层次信息
在SMCRA的所有层次水平,你还可以提取一个 完整id 。完整id是包含所有从顶层对象(结构)到当前对象的id的一个元组。一个残基对象的完整id可以这么得到:
full_id = residue.get_full_id()
print full_id
("1abc", 0, "A", ("", 10, "A"))
这对应于:
id为”1abc”的结构
id为0的模型
id为”A”的链
id为(” “, 10, “A”)的残基
这个残基id表示该残基不是异质残基(也不是水分子),因为其异质值为空;而序列标识符为10,插入码为”A”。
structure中获取model
结构域对象的id是一个整数,源自该模型在所解析文件中的位置(自动从0开始)。晶体结构通常只有一个模型(id为0),而NMR文件通常含有多个模型。然而许多PDB解析器都假定只有一个结构域, Bio.PDB 中的 Structure 类就设计成能轻松处理含有不止一个模型的PDB文件。
structure.get_models()
## 或者
first_model = structure[0]
从structure中获取chains
chains=structure.get_chains()
得到的chains为chains类对象,它包含该蛋白质所有肽链。
print(list(chains))
'''------------------'''
>>> [<Chain id=A>, <Chain id=B>]
从structure/chain中获取residues
从structure、chain中获取residues方法是相同的,structure 是从整体蛋白质获取残基,chain是指定某条链获取残基。
chains=structure.get_chains()
chain_A=list(chains)[0]
residues_A=chain_A.get_residues()
以A链为例,把chains转成列表,列表中第一个表示A链对象,之后用get_residues()函数即可。
若从structure获取所有残基,用下面方法即可。
residues=structure.get_residues()
将residues转成列表,列表中每个值为一个残基类,包含该残基相关信息。
一个残基id是一个三元组:
异质域 (hetfield),即:
‘W’ 代表水分子
‘H_’ 后面紧跟残基名称,代表其它异质残基(例如 ‘H_GLC’ 表示一个葡萄糖分子)
空值表示标准的氨基酸和核酸
序列标识符 (resseq),一个描述该残基在链上的位置的整数(如100);
插入码 (icode),一个字符串,如“A”。插入码有时用来保存某种特定的、想要的残基编号体制。一个Ser 80的插入突变(比如在Thr 80和Asn 81残基间插入)可能具有如下序列标识符和插入码:Thr 80 A, Ser 80 B, Asn 81。这样一来,残基编号体制保持与野生型结构一致。
因此,上述的葡萄酸残基id就是 (’H_GLC’, 100, ’A’) 。如果异质标签和插入码为空,那么可以只使用序列标识符:
print(list(residues))
'''--------------------'''
>>> [<Residue TYR het= resseq=3 icode= >,
<Residue LYS het= resseq=4 icode= >,
<Residue ASN het= resseq=5 icode= >,.....]
其中Residue表示氨基酸名;het是异质域,'W’表示水,'H_残基名’表示非标准氨基酸,空表示标准氨基酸;resseq为氨基酸序列编号;icode为插入码。
对于每个残基,使用get_resname()可获取残基名,使用get_atoms()函数可以获取该residue中所有原子。
resname=list(residues)[0].get_resnames()
atoms=list(residues)[0].get_atoms()
上述代码第一行获取第一个氨基酸残基名,第二行获取氨基酸残基所有原子。
residue.get_resname() # returns the residue name, e.g. "ASN"
residue.is_disordered() # returns 1 if the residue has disordered atoms
residue.get_segid() # returns the SEGID, e.g. "CHN1"
residue.has_id(name) # test if a residue has a certain atom
print(list(atoms))
'''------------------'
>>>[<Atom N>, <Atom CA>, <Atom C>, <Atom O>, <Atom CB>,
<Atom CG>, <Atom CD1>, <Atom CD2>, <Atom CE1>,
<Atom CE2>, <Atom CZ>, <Atom OH>]
residue中其它方法:
get_segid:返回segidget_unpacked_list:undisordered原子组成的列表。sort():将原子排序,N,CA,C,O等排在前面。is_disordered():判断氨基酸中十分含有disordered原子,若含有则返回True。
从structure/chain/residues中获取原子
从residues中获取atoms方法前面已讲述,从structure、chain中获取原子信息方法是与前面相同的,仍使用get_atoms()函数。
atoms=structures.get_atoms()
atoms_chain_A=chain_A.get_atoms()
从atoms中获取原子信息
子对象储存着所有与原子有关的数据,它没有子类。原子的id就是它的名称(如,“OG”代表Ser残基的侧链氧原子)。在残基中原子id必需是唯一的。此外,对于紊乱原子会产生异常,见 11.3.2 小节的描述。
原子id就是原子名称(如 ’CA’ )。在实践中,原子名称是从PDB文件中原子名称去除所有空格而创建的。
但是在PDB文件中,空格可以是原子名称的一部分。通常,钙原子称为 ’CA…’ 是为了和Cα原子(叫做 ’.CA.’ )区分开。在这种情况下,如果去掉空格就会产生问题(如统一个残基中的两个原子都叫做 ’CA’ ),所以保留空格。
在PDB文件中,一个原子名字由4个字符组成,通常头尾皆为空格。为了方便使用,空格通常可以去掉(在PDB文件中氨基酸的Cα原子标记为“.CA.”,点表示空格)。为了生成原子名称(然后是原子id),空格删掉了,除非会在一个残基中造成名字冲突(如两个原子对象有相同的名称和id)。对于后面这种情况,会尝试让原子名称包含空格。这种情况可能会发生在,比如残基包含名称为“.CA.”和“CA…”的原子,尽管这不怎么可能。
所存储的原子数据包括原子名称,原子坐标(如果有的话还包括标准差),B因子(包括各向异性B因子和可能存在的标准差),altloc标识符和完整的、包括空格的原子名称。较少用到的项如原子序号和原子电荷(有时在PDB文件中规定)也就没有存储。
为了处理原子坐标,可以用 ’Atom’ 对象的 transform 方法。用 set_coord 方法可以直接设定原子坐标。
a.get_name() # atom name (spaces stripped, e.g. "CA")
a.get_id() # id (equals atom name)
a.get_coord() # atomic coordinates
a.get_vector() # atomic coordinates as Vector object
a.get_bfactor() # isotropic B factor
a.get_occupancy() # occupancy
a.get_altloc() # alternative location specifier
a.get_sigatm() # standard deviation of atomic parameters
a.get_siguij() # standard deviation of anisotropic B factor
a.get_anisou() # anisotropic B factor
a.get_fullname() # atom name (with spaces, e.g. ".CA.")
get_vector():原子坐标的vector。get_coord():原子坐标。get_fullname():原子全名。get_serial_number() or serial number():原子序号(从1开始)。get_sigatm():原子坐标标准差。get_bfactor():bfactor温度因子。set_?:设置属性,'?'可以为coord、bfactor、serial_number等。
从结构中提取指定的 Atom/Residue/Chain/Model
model = structure[0]
chain = model['A']
residue = chain[100]
atom = residue['CA']
## 还可以用一个快捷方式:
atom = structure[0]['A'][100]['CA']