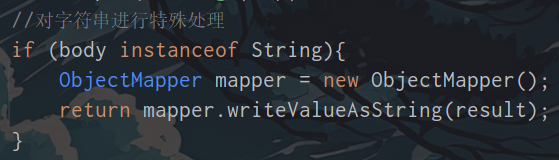
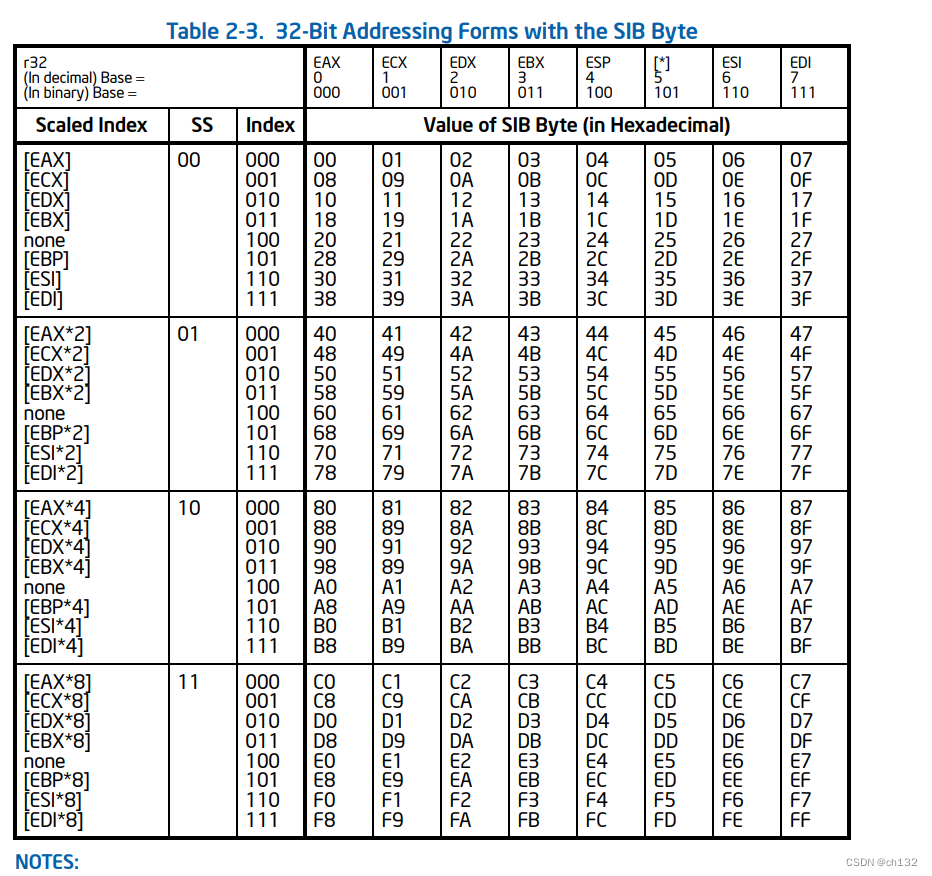
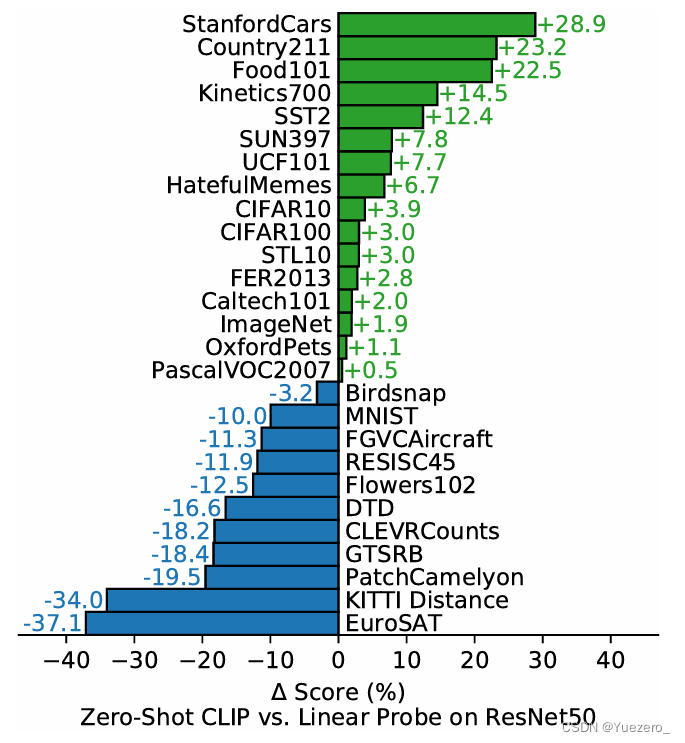
新手在学习DFT计算时,在熟悉了基本的操作和VASP输入文件后,首先就会学习到结构优化、自洽计算和能带的计算。
而笔者学习DFT计算这些年来看到太多新手学者踩到大大小小的坑,其中能带看起来不连续或者能带不连续则是几乎必踩的坑之一。
这些初学者大多使用了vaspkit的303功能来一键生成了能带的高对称点路径文件KPATH.In文件并复制成了KPOINTS进行能带计算并同样的通过vaspkit的211和21系列的其它功能进行能带处理。
vaspkit软件作者专门写了一个推送来介绍绘制图片时能带不连续的情况。能带看起来不连续,怎么办?
产生的能带基本都是稳定连续的,当然也有假装连续的,出现断层的情况也是很常见。
石墨烯能带(vaspkit绘图)
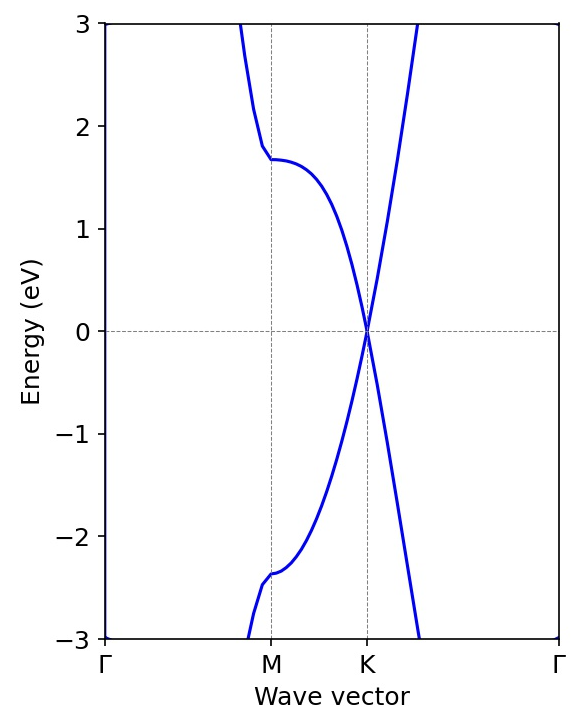
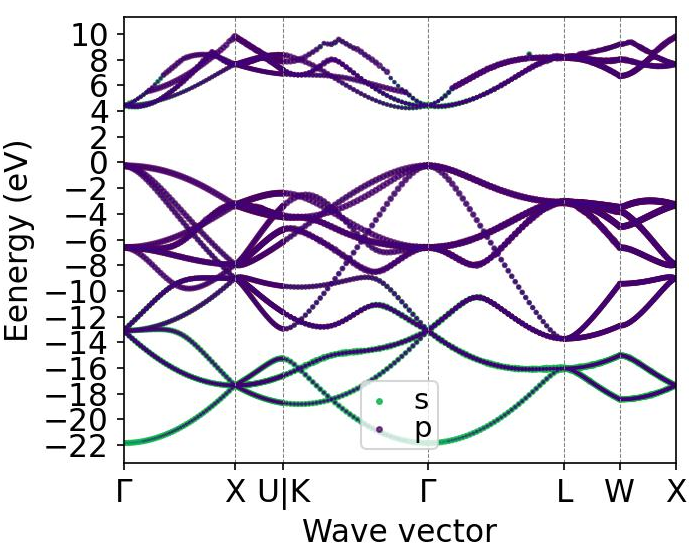
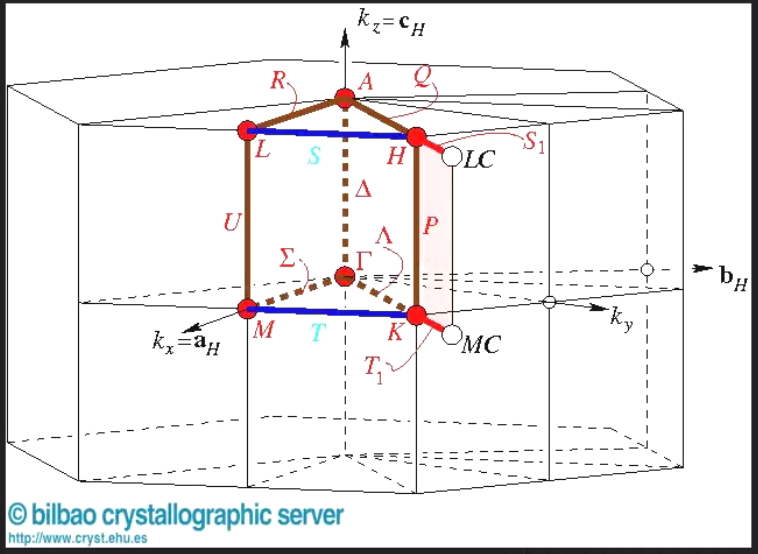
在笔者前期计算的案例(本期推送重新发表:掺杂三个原子后能带不连续了,何解,旧版链接“掺杂三个原子后能带不连续了?” 何解?)中计算了类金刚石模型构模型的能带结构,从中可以看到能带明显是连续的,但是在高对称点路径的写法上存在一个奇怪的地方就是”U|K“,这是因为在高度对称的原胞的布里渊区中,U点(0.625 0.25 0.625)和K点(0.375 0.375 0.25)所处能量状态是几乎等同的,所以看起来连续在一起。
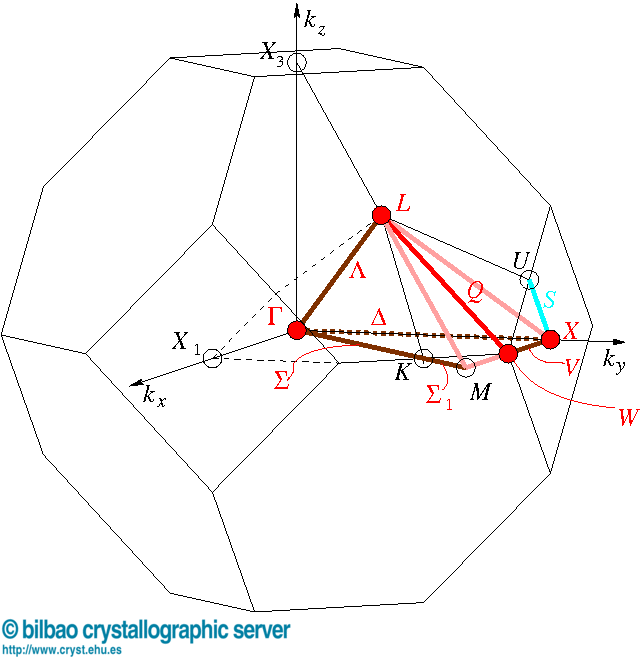
对于该空间群的布里渊区来说,如下图所示,K点位于两个六边形表面共线的中点,而U点则为六边形和四边形表面共线的中点,高度对称的结构中所以存在等同的能力值
采用vaspkit产生能带路径时,则会直接产生不连续的路径。

这里我们介绍一下计算能带路径的写法,如上图,此KPOINTS为line-mode模式,在第一个高对称点GAMMA(0 0 0)后接上了另一个高对称点X(0.5 0 0.5),然后中间空一行,这就是能带路径中Γ-X的路径的k点,而在此之后,留一行空白,再以高对称点X开头,后接另一个高对称点U(0.625 0.25 0.625),则是在前面Γ-X之后再接一段X-U的能带,因为前一段的末尾是X点,后一段的开头也是X点,那么能带则一定会连续在一起。
而在U点后空了一行后,再开头的不是U点,而是K点(0.375 0.375 0.25),那么则如果U点和K点的能量值不等同的话,能带则会出现断点,前一段X-U和后一段K-Γ则无法连续在一起。比如在之前的案例中掺杂之后的能带图

后面其他能带的高对称点的写法暂不赘述。
修改方法也很简单,首先不考虑布里渊区内高对称点连续性的话,可以在KPOINTS的能带路径中加上U-K的这一段路径,如下所示。
K-Path Generated by VASPKIT.20Line-ModeReciprocal0.0000000000 0.0000000000 0.0000000000 GAMMA0.5000000000 0.0000000000 0.5000000000 X0.5000000000 0.0000000000 0.5000000000 X0.6250000000 0.2500000000 0.6250000000 U0.6250000000 0.2500000000 0.6250000000 U0.3750000000 0.3750000000 0.7500000000 K0.3750000000 0.3750000000 0.7500000000 K0.0000000000 0.0000000000 0.0000000000 GAMMA0.0000000000 0.0000000000 0.0000000000 GAMMA0.5000000000 0.5000000000 0.5000000000 L0.5000000000 0.5000000000 0.5000000000 L0.5000000000 0.2500000000 0.7500000000 W0.5000000000 0.2500000000 0.7500000000 W0.5000000000 0.0000000000 0.5000000000 X
依照此路径计算得到的NaCl能带结构如下图所示,多出来了U-K点的能带路径,由于二者能量相近,路径上不存在太大的能量波动,整一段显得较为平缓。

当然,如果想正确分析能带结构,则需要深入学习和理解布里渊区和高对称点的含义,并合理的设置能带的路径。
下面是最近一个初学者与笔者讨论的例子,也希望给大家提供经验。
最初呈现不连续的能带结构,如下图,最右边的部分,还没有标明高对称点的位置但是可以很明显的看出有曲折。

最开始使用的KPOINTS文件以及笔者建议他修改的位置(通过横线标出)
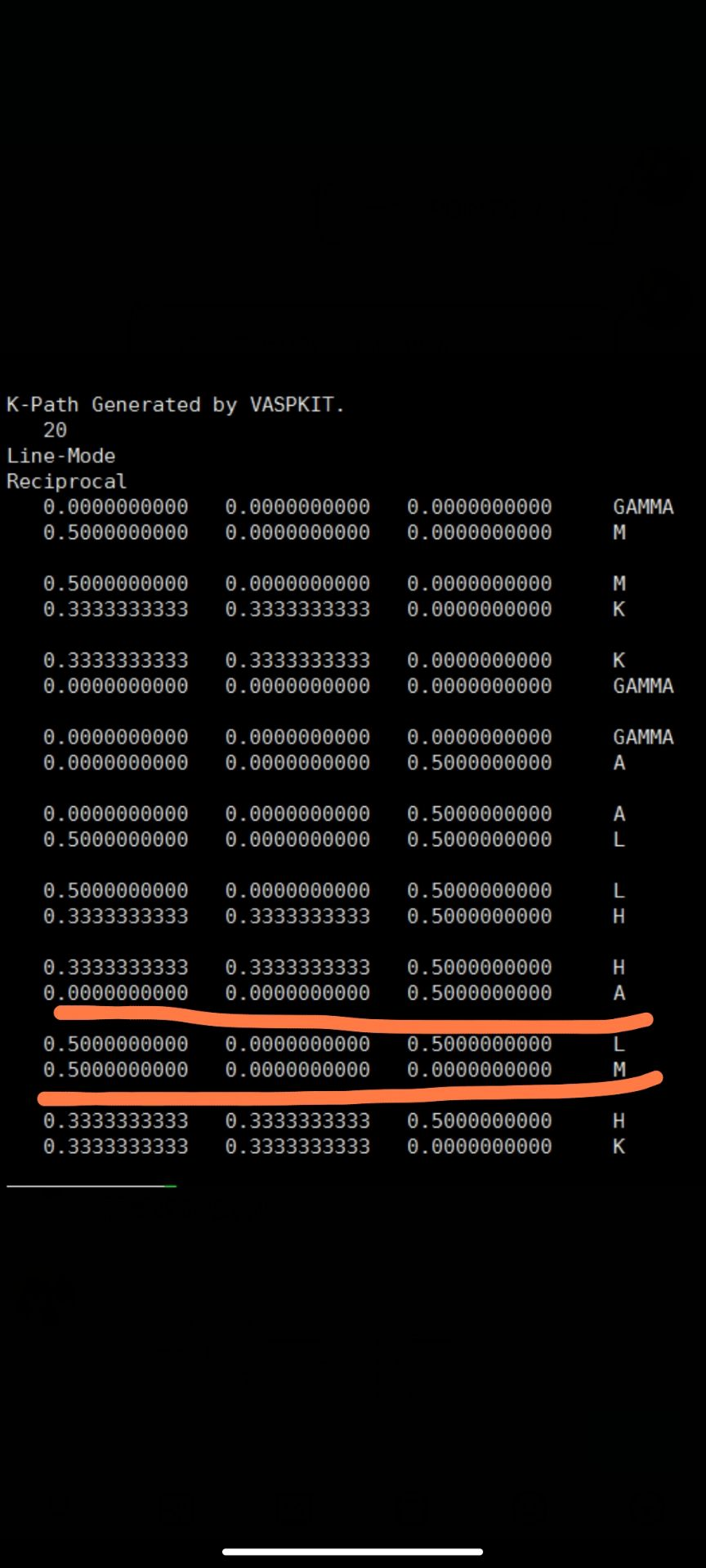
根据空间群对称性查到的合适的高对称点的信息
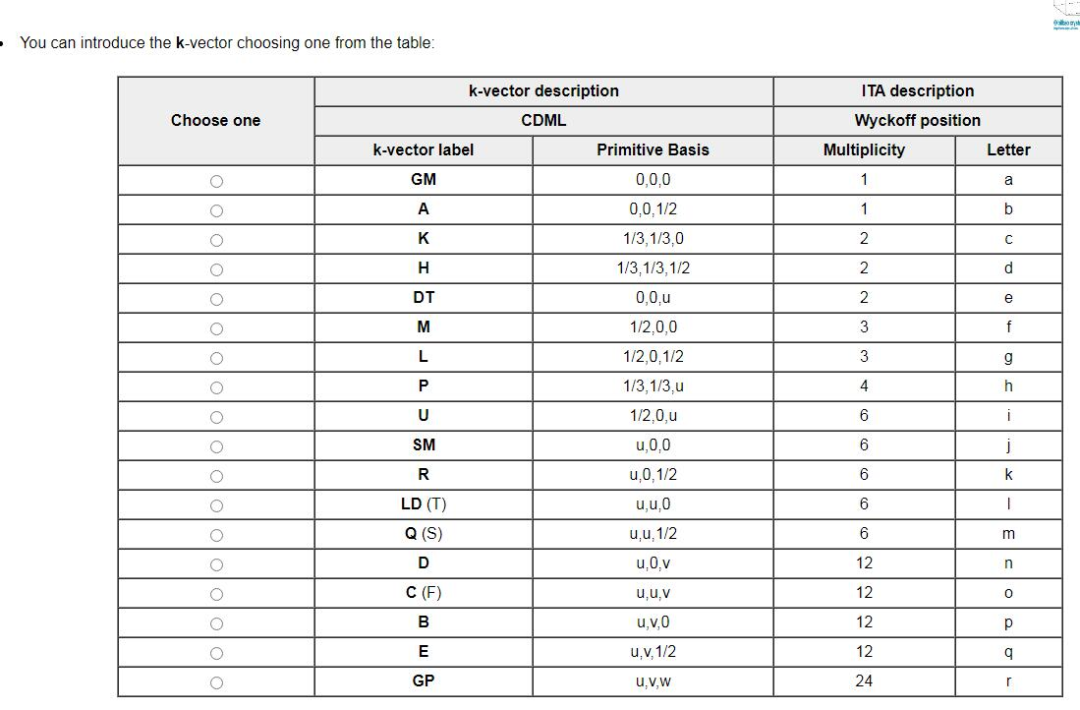
更改后的能带路径

以及重新计算的能带结构