目录
- 蛋白质结构预测
- 三种方法
- 同源建模(比较建模)
- 穿线法
- 从头预测(ab initio)
- 基于假设
- 推荐策略
- 精度与方法选择
- Alphafold2相关信息
蛋白质结构预测
三种方法
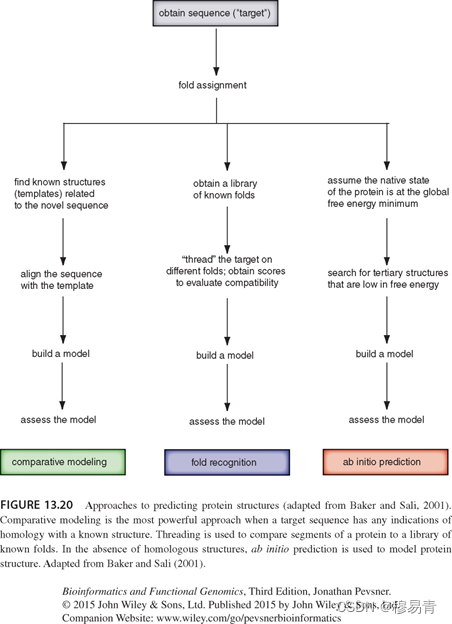
同源建模(比较建模)
建模4步骤
1.模板选择和确定折叠构象
通过blast或delta-blast搜索同源蛋白质序列或结构,识别保守和可变区域。
常用查询数据库:
PDB数据库:https://www.rcsb.org/
CATH:http://cathdb.info/
SCOP:https://scop.mrc-lmb.cam.ac.uk/
2.把目标蛋白质和模板蛋白质进行比对
一般序列一致性大于50%。或比对到序列区域足够长,说明这两个蛋白质可能有相似结构,精度较高。
3.建立结构模型
4.对结构模型进行评估
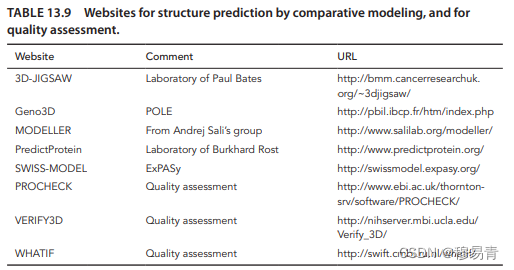
评估工具
VERIFY3D: http://nihserver.mbi.ucla.edu/Verify_3D/
PROCHECK:http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/
CMBI-WHATIF:http://swift.cmbi.ru.nl/whatif/
在线建模服务网站
SWISSMODEL: http://swissmodel.expasy.org/
MODELLER: http://www.salilab.org/modeller/
PredictProtein: http://www.predictprotein.org/
穿线法
输入的目标序列被打断成一个个片段,然后在一个已知折叠的模板库上进行“穿线”。打分函数会评估目标序列与已知结构之间的相容性。
从头预测(ab initio)
基于假设
氨基酸序列包含了关于蛋白质结构的所有信息;球蛋白会折叠成自由能最低的结构。
推荐策略
Rosetta
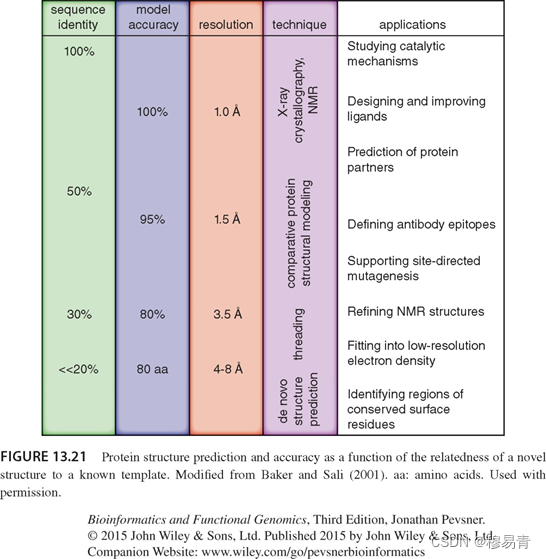
精度与方法选择
Alphafold2相关信息
[AlphaFold2 讲解(2) - 知乎 (zhihu.com)](https://zhuanlan.zhihu.com/p/464269311)
[AlphaFold2算法详解_李划水员的博客-CSDN博客_alphafold2原理](https://zhuanlan.zhihu.com/p/464269311)
[【公开课】基于AI预测蛋白质折叠的三维空间结构——AlphaFold2原理及安装使用_哔哩哔哩_bilibili](https://www.bilibili.com/video/av504954705?zw&vd_source=999ee47504687d8fa8e31dc1363002ca)