使用蛋白ID如何进行KEGG和GO富集分析 - 知乎 (zhihu.com)
昨天,有个童鞋咨询如何使用蛋白ID进行功能富集分析,功能富集分析主要是KEGG和GO。

思路
- 蛋白ID转UniProt数据库ID
- UniProt数据库ID转KEGG和GO号
- 使用KEGG和GO号进行富集分析
教程(实操开始)
蛋白ID数据类型
蛋白ID的数据是的使用;进行隔分的,如果要整理成一列数据,我最开始想到的就是使用sed进行处理。

「注:个人还是建议使用fa序列进行mapping,但是只要获得正确的结果,也无所谓。」
1. 蛋白ID转UniProt数据库ID
使用UniProt数据库的工具UniProtKB ID Mapping(https://www.uniprot.org/uploadlists/)
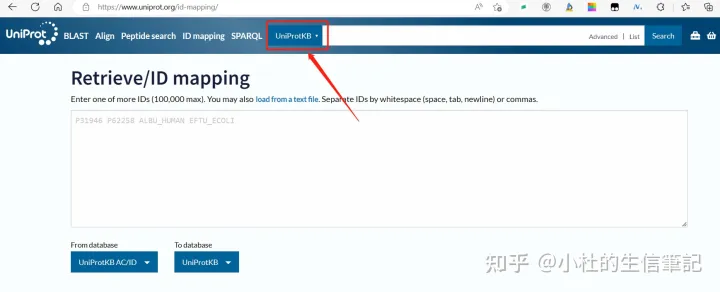
「1. 直接将数据复制-粘贴到过来即可,无需操作。」

「2. 选择UniProtKB AC/ID To KEGG Pathway,如下图所示。」

「3. 选择好后直接点解Map即可。」

等待一段时间,即可完成Completed

4. Download数据

格式选择其中一种即可。
获得结果
目前,已经获得UniProt数据库ID。那么,我们可以直接试用其进行转换即可,方法有两种,一种是直接在KEGG数据库中进行转换,一种是使用云平台进行转换。
- KEGG数据库中机芯富集https://www.genome.jp/kegg/mapper/color.html,自己做了没成功。 2.使用云平台进行转换(我们的童鞋使用基迪奥云平台进行转换,获得如下结果)

「依旧是你喜欢的样子,GO号和KEGG号都有,可以直接使用。」
方式二: 使用R语言进行转换
我们在这里尝试很多方式,依旧是没有成功!需要同学们的帮助,如果你有好的建议或方式,欢迎进行交流,这个问题一直留个大家讨论!!!!
代码一
安装R包
install.packages("UniprotR")
install.packages("KEGGREST")
install.packages("clusterProfiler")
##
# 如果安装不了换成BiocManager::install()安装
##'@加载
library(UniprotR)
library(KEGGREST)
library(clusterProfiler)加载数据
##'@加载蛋白ID数据
protein_ids <- read.table("test.02.txt",header = F)
#protein_ids <- sapply(protein_ids, as.character)
protein_ids <- as.character(protein_ids$V1)
head(protein_ids)
> head(protein_ids)
[1] "N1PAF1" "G2WAT7" "C8Z5A3" "C7GN09" "A6ZYE7" "A0A815XJ81"
### 转换
kegg_ids <- idmapping(protein_ids, from = "ACC",
to = "KEGG_ID", format = "list",
verbose = FALSE)$KEGG_ID
这里报错,找不到"idmapping"

报错后后面的依旧是进行不了,找了很多教程依旧是没找到。
代码二
这里依旧是同样的问题,那么大家看一下代码吧。我个人觉得,这个代码的可靠性更高一些。这里使用org.Sc.sgd.db包。
加载R包
library(clusterProfiler)
library(org.Sc.sgd.db)
library(pathview)
library(enrichplot)加载数据
##'@加载蛋白ID数据
protein_ids <- read.table("test.02.txt",header = F)
#protein_ids <- sapply(protein_ids, as.character)
protein_ids <- as.character(protein_ids$V1)
head(protein_ids)
转换
kegg_ids <- idmapping(protein_ids, "ACC", "KEGG_ID", format = "list", "org.Sc.sgd.db")
代码三
加载R包
install.packages("KEGGREST")
install.packages("org.Sc.sgd.db")
library(KEGGREST)
library(org.Sc.sgd.db)加载数据-(同上)
转换
kegg_ids <- vector("list", length(protein_ids))
for(i in seq_along(protein_ids)){
kegg_ids[[i]] <- tryCatch(keggConv(protein_ids[i],
"ncbi-proteinid", "pathway+gene"),
error = function(e) NA)
}
kegg_ids <- unlist(kegg_ids[!is.na(kegg_ids)])
「没有结果!!!!」
「由于没有结果,后面富集分析也就是不能继续了!!」
「如果你想折腾,可以继续折腾!!」
「如果,你不想折腾,也开始直接使用第一种方法即可!」
「往期文章:」 「1. 最全WGCNA教程(替换数据即可出全部结果与图形)」 WGCNA分析 | 全流程分析代码 | 代码一
WGCNA分析 | 全流程分析代码 | 代码二
WGCNA分析 | 全流程代码分享 | 代码三
「2. 精美图形绘制教程」
精美图形绘制教程
![[C++] C++入门第一篇 -- 命名空间,输入输出,缺省函数,函数重载底层原理](https://img-blog.csdnimg.cn/img_convert/fd87802f510b5c3cf790cd01cf140dce.png)