KEGG COMPOUND 数据库 - 简书 (jianshu.com)
kegg id号转换为可读 的name :使用KEGGREST
#-------------kegg id的entry和Name转换 https://zhuanlan.zhihu.com/p/545494092
#BiocManager::install("KEGGREST") #安装KEGGREST这个包
library(KEGGREST) #加载该R包
listDatabases() #查看可以利用的数据库
keggList()
##获取pathway(所有物种)数据集中的数据
pathway<- keggList("pathway")
head(pathway)
rat_pathway=keggList("pathway",organism = "rno")
head(rat_pathway)
#hsa_pathway <- keggList("pathway","hsa") # 获取KEGG数据库中所有人类通路 https://zhuanlan.zhihu.com/p/434383719
hsa_pathway=rat_pathway
hsa_path <- data.frame(hsa_pathway) # 转成数据框,方便后续分析
print(head(hsa_path))
hsa_path$pathID <- substr(rownames(hsa_path),6,nchar(rownames(hsa_path)[1])) # 提取pathway ID
kegg compound id 与HMDB转换 :使用metabolystr包
{
0. #kegg id
library(KEGGREST) #加载该R包
library(tibble)
listDatabases() #查看可以利用的数据库
keggList()
##获取pathway(所有物种)数据集中的数据
pathway<- keggList("pathway")
head(pathway)
rat_pathway=keggList("pathway",organism = "rno")
rat_pathway=data.frame(rat_pathway) %>%rownames_to_column(var = "kegg entry id")
rat_pathway$metabolic_pathway=str_split(rat_pathway$rat_pathway,pattern = " - Rattus norvegicus \\(rat\\)",simplify = T)[,1]
print(head(rat_pathway))
save.mat$metabolic_pathway=rownames(save.mat)
print(head(save.mat))
# Merge the data frames based on the "metabolic_pathway" column
merged_data <- merge(save.mat, rat_pathway, by = "metabolic_pathway")
# Print the first few rows of the merged data
print(head(merged_data))
1. #kegg compound id
rm(mSet)
mSet<-InitDataObjects("list", "msetora", FALSE)
cmpd.vec<-tmp.vec
mSet<-Setup.MapData(mSet, cmpd.vec);
mSet<-CrossReferencing(mSet, "name");
mSet<-CreateMappingResultTable(mSet)
metabolite_hmdb_kegg=mSet[["dataSet"]][["map.table"]] %>% as.data.frame()
metabolite_hmdb_kegg=metabolite_hmdb_kegg[ metabolite_hmdb_kegg$KEGG!="NA"&
!is.na(metabolite_hmdb_kegg$KEGG),]
print(head(metabolite_hmdb_kegg))
dim(metabolite_hmdb_kegg)
2.#uniport id
deg_proteins=read.csv("/home/data/t040413/wpx/wpx_proteinomics/1_model_success_3/LCT(14+28)-NT-D28-Normal_control _differential_proteins.csv")
deg_proteins=deg_proteins[deg_proteins$regulate!="NOT",]$protein_name
print(getwd())
print(gene.idtype.list )
data(rn.list);
print(names(rn.list))
gene.ensprot <- sim.mol.data(mol.type = "gene", id.type = gene.idtype.list[4])
head(gene.ensprot)
cpd.simtypes
head(deg_proteins$protein_name)
print(head(metabolite_hmdb_kegg$KEGG))
head(merged_data)
pv.out <- pathview(gene.data = deg_proteins$protein_name,
cpd.data = metabolite_hmdb_kegg$KEGG,
gene.idtype = "UNIPROT", cpd.idtype = "kegg",
pathway.id = "rno01040", # merged_data[,"kegg entry id"] [1],
species = "rno", out.suffix = "sel.genes.sel.cpd",
keys.align = "y",
kegg.native = T,
key.pos = demo.paths$kpos1[1],
limit = list(gene = 1, cpd = 1),
bins = list(gene = 1, cpd = 1),
na.col = "gray", discrete = list(gene = T, cpd = T))
pv.out <- pathview(gene.data = deg_proteins$protein_name,
cpd.data = metabolite_hmdb_kegg$KEGG,
pathway.id = "rno01040",
gene.idtype = "UNIPROT", cpd.idtype = "kegg",
species = "rno",
out.suffix = "sgfssel.genes.sel.cpd ",
keys.align = "y", kegg.native = T,
key.pos = demo.paths$kpos1[i],
limit = list(gene = 5, cpd = 2),
bins = list(gene = 5, cpd = 2),
na.col = "gray", discrete = list(gene = T, cpd = T))
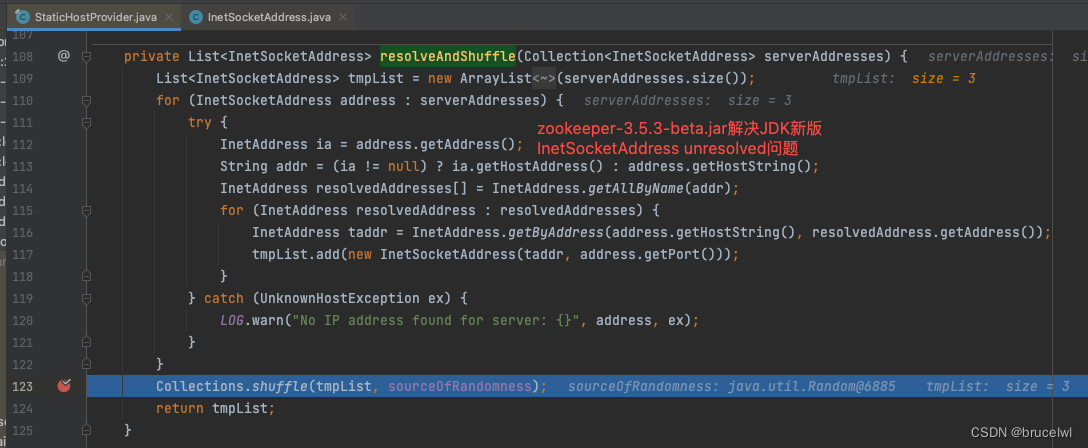
}kegg compound 数据库存储了在生命活动中发挥作用的各种小分子,生物大分子和其他类型的化学物质,采用C number 进行标识,比如C00047, 代表L-赖氨酸。除了名称等信息外,还存储了该物质的化学结构和其他相关信息;
对于所有compound 的分类详见 Brite 数据库
image
Module 是ko的集合,但是ko只是基因集,真正参与生命活动的是这些基因的产物,在产物发挥作用的时候,也需要compound 的参与,所有会给出compound 相关的module。
Enzyme 数据库保存各种酶的相关信息,酶作为催化剂调控一些生物学过程的发生和进行,在这个过程中肯定也会有compound 的参与;比如1.1.1.306 这种酶催化的反应中, 供体提供甲酸才能进行反应,所以会给出compound 对应的Enzyme 编号;
总结
compound 数据库存储了参与生命活动的各种分子的信息,数据库中的记录用C Number唯一标识, 每条分子都有对应的化学式,结构式,分子量等基本信息;
compound 和reaction , module, pathway, enzeme 等多个数据库都有联系;