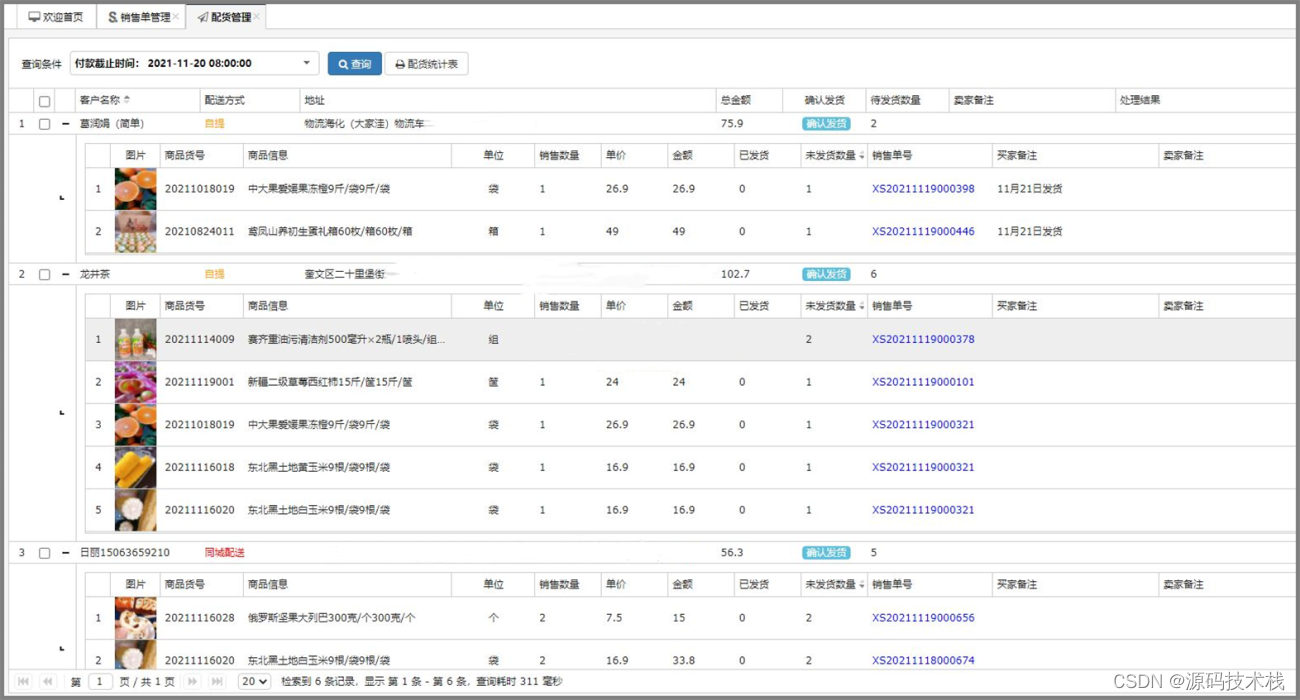
计算背景:
利用高分子有机物等活性材料对有毒分子、原子、离子在真空、水溶液、有机溶液等环境下吸附,已是当今环境科学、矿物学、土壤化学等学科领域研究的热点。但如何确定最佳吸附位点以计算其吸附能就显得尤为重要。
现阶段多数物质的吸附均依据粒子间的静电吸附靠近,从而得到进一步的稳定吸附。如何确定活性材料的吸附位点难以从常规测试中得到,EDS测试难以识别具有相同元素的不同吸附位点,因此通过计算机模拟计算化学物质的静电势来确定吸附位点以及计算结合能是一个非常好的选择。
主要步骤:
(1) 利用GaussView或其他软件绘制出待计算物质的分子模型(以阳离子树脂为例);
(2) 选择合适的泛函、基组以及溶剂环境进行结构优化,同时务必记得要保留chk文件(要不然结果中没有静电势)
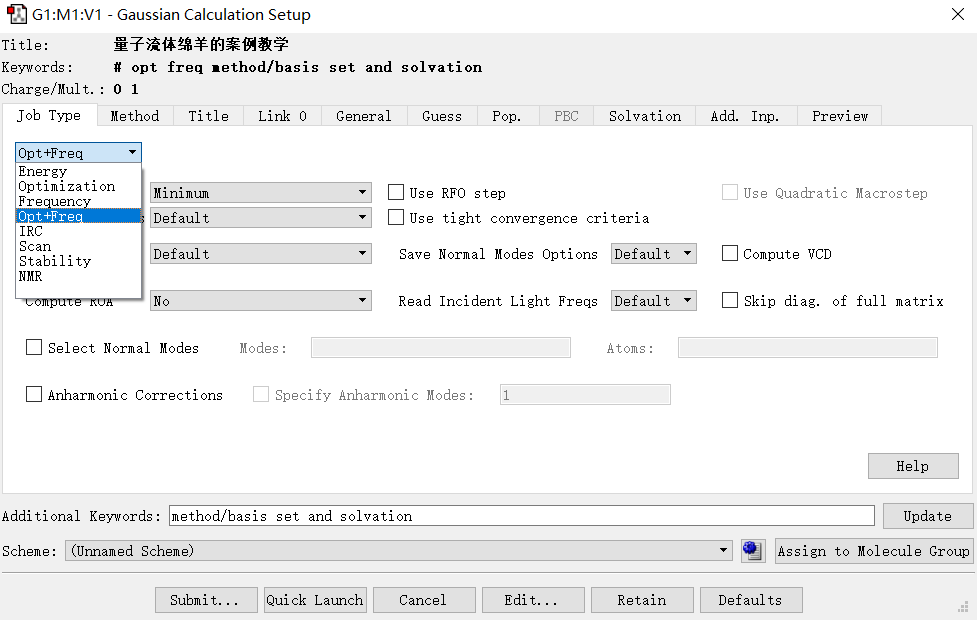
(3) 使用GaussView打开计算完成后的chk/fchk/fch文件,右键Results→Surface/Contours…

在Cube Available一栏中选择Cube Action→New Cube

结果计算完毕后在Surfaces Available中选择New Mapped Surface,在弹出窗口中选择ESP,点击ok,依据体系原子个数和电脑显卡能力不同,等待几秒到几分钟不等。(MO和Dencity可能需要依据需求进行修改)

(4) 对结果的可视化方式进行合适修改后,一个漂亮的静电势图片就出来了,依据上方的比例尺可以看出红色是负电,蓝色是正电,比例尺极值数值可以修改,图片颜色也会相应变化。
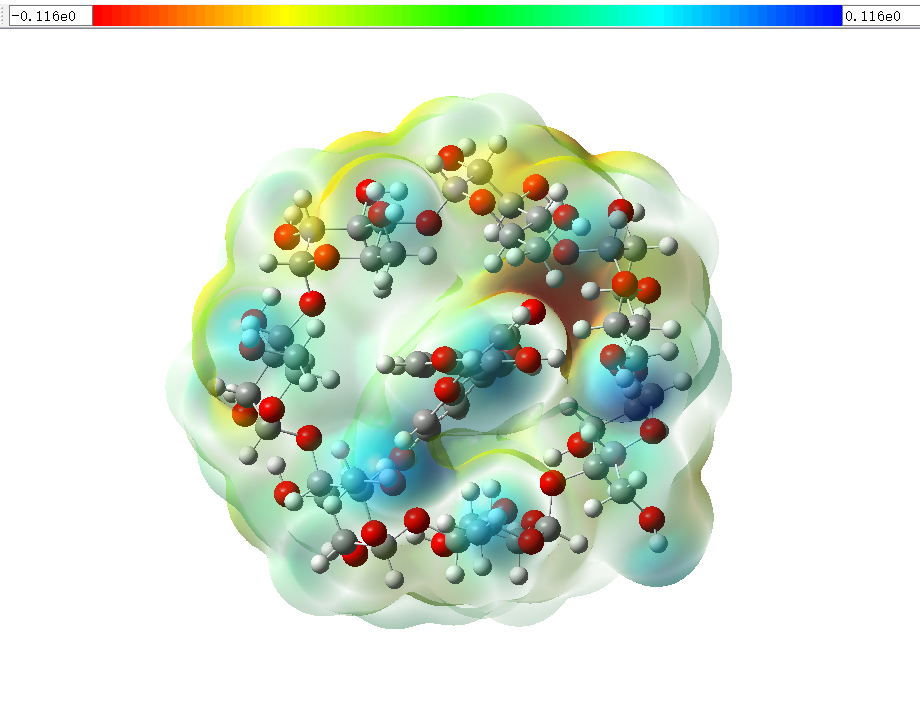
(5) 我们的三价Cr离子带正电,所以倾向于吸附在图中红色(负电)的部分,具体位点可在Gaussian默认储存文件位置找到.cub 文件,里面会列出几个较正和较负位点的坐标以及值。
总结:
将电荷极小值坐标复制到gjf文件中,赋予该坐标Cr原子,打开后适当调整即可。依据最佳吸附位点的计算,可对后续材料改良、新材料制备等进行预测,对后续实验具有指导性意义。
欢迎大家关注我们的公众号"320科技工作室"联系我们.

![Yocto系列讲解[驱动篇]89 - 内核通知事件notifier chain驱动示例](https://img-blog.csdnimg.cn/f64e1f114d7045eaa57a680daec4395b.png)


![[附源码]JAVA毕业设计心理学网站(系统+LW)](https://img-blog.csdnimg.cn/90afb46962664607818512323224633d.png)