这里,介绍一下BGEN格式的数据,他的文件格式是这样的:a.bgen,这是一个新的数据格式,目前应用不如plink的二进制文件:.bim,.bed,.fam。这里介绍一下如何相互转换。
1. bgen格式介绍
现代遗传关联研究通常使用数万至数十万个体的数据,这些数据是全基因组数千万标记的基因分型或估算的。基于这些数据的文本表示的传统数据格式(如IMPUTE输出的GEN格式或变量调用格式)有时不太适合这些数据量。事实上,对于简单的程序,解析这些格式所花费的时间可以支配程序执行时间。
本页介绍了二进制GEN文件格式(“BGEN”格式),旨在解决这些问题。BGEN是一种稳健的格式,其设计具有特定的混合特性,我们认为这对此类研究很有用。它的目标是用于大型、潜在的遗传数据集。主要功能包括:
-
存储直接输入和输入数据的能力。
-
存储非阶段基因型和阶段性单倍型数据的能力。
-
通过使用高效的、可变精度的压缩位表示和压缩,文件大小较小。
-
每变量压缩的使用使格式易于索引和编目。
例如,下图显示了在1号染色体上121668个SNP的18496个样本的数据集中,列出各种常见格式(Y轴)、文件大小(X轴)的变体识别数据(即基因组位置、ID字段和等位基因)所需的时间。下面定义的BGEN的两个变体都显示了出来。
下面图中,X周是文件的带下,用log10转换了。Y坐标是处理的时间,文件的格式:
- 压缩的gen文件
- 压缩的vcf文件
- bgen文件
- bed文件
- vcf原始文件
- gen文件
可以看到,bgen1.2,bed,bgen1.1三个格式,文件大小最小,处理时间最短,bed文件没有bgen包含的信息多,所以,这里推荐使用bgen格式。
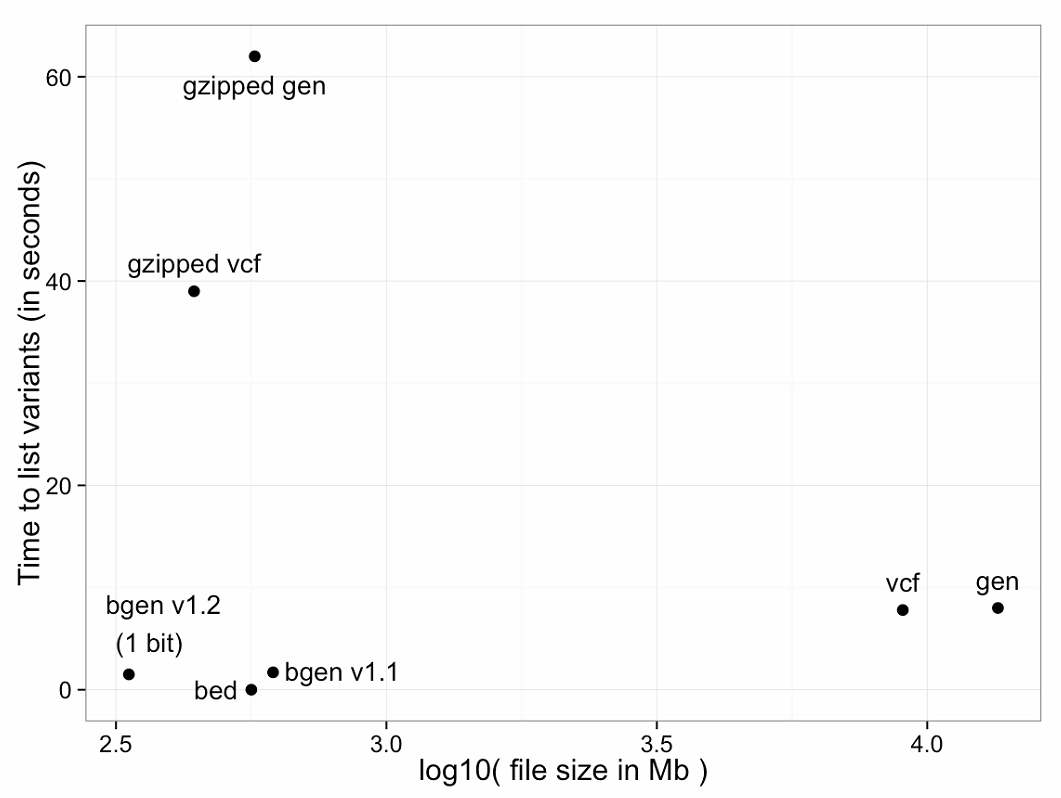
对于PLINK二进制(.bid)文件,标识数据存储在单独的文件(.bim文件)中,因此时间实际上为零。对于基于文本的格式,文件压缩的使用和读取性能之间存在显著的权衡。BGEN以334Mb存储了22.5亿个基因型的整个数据集,每个基因型略多于一位,在该测试中耗时1.5秒。
(当然,所有格式的性能优化都是可能的,因此上面的图不会代表最佳可能的时序,但应被视为说明性的。)
BGEN格式已用于多个主要项目,包括Wellcome Trust Case Control Consortium 2、MalariaGEN项目和ALSPAC研究。它已被英国生物银行采用为全基因组估算基因型的发布格式。
2. 处理bgen的软件
这里,常用的软件:
- Mega2
- LDstore
- PLINK
- STITCH

对于R语言用户,可以用:rbgen包处理
对于Python用户,可以用:bgen-reader和pybgen包处理
还有一些C++的程序可以处理,具体参考:https://www.well.ox.ac.uk/~gav/bgen_format/software.html
3. bgen格式转为plink的文件(ped,map)
注意,plink读取bgen文件时,需要指定:
- .bgen
- .sample
这两个文件都要存在。bgen文件是二进制文件,sample文件是包括ID_1,ID_2, missing sex的四列数据。
plink2 --bgen t1.bgen 'ref-last' --sample t1.sample --export ped --out x1
- –bgen文件:指定t1.bgen,后面跟着参数:
ref-last,表示ref是放到后面,而不是默认的major为ref - –sample 文件,指定 t1.sample,后面跟着sample文件,这两个文件要分开指定
- –export ped,指定输出的格式,是输出plink的文本文件
4. bgen格式转为plink的二进制文件(bed,bim和bed)
plink2 --bgen t1.bgen 'ref-last' --sample t1.sample --make-bed --out x1
参数和上面一致,输出用--make-bed定义输出格式为bed,bim,fam。
5. plink二进制文件转为bgen格式
plink2 --bfile a1 --export bgen-1.1 --out t1
- –bfile,读取二进制的前缀
- –export bgen-1.1 输出bgen格式的文件,还可以用bgen-1.2
就酱!
其它参考资料:plink2.0和plink1.9的忧伤笔记
![[附源码]计算机毕业设计JAVA中小企业人事管理系统](https://img-blog.csdnimg.cn/8a97c84c88684ba5b41554b79950a139.png)