编者按:近年来,深度学习技术在分子微观结构预测中取得了巨大的进展。然而,分子的宏观属性和功能往往取决于分子结构在平衡态下的分布,仅了解分子的微观结构还远远不够。在传统的统计力学中,分子动力学模拟或增强采样等是获得平衡分布中采样的常用方法,但这些方法昂贵又耗时。
针对这个长期且艰巨的挑战,微软研究院发布了可用于预测分子结构平衡分布的深度学习框架 Distributional Graphormer (DiG)。DiG 可以快速生成真实多样的构象,进而为实现从单一结构预测到平衡分布预测的突破奠定基础。实验表明,DiG 在蛋白质、蛋白质-配体复合物和催化剂-吸附质系统等采样任务中,展现出了优异的性能和潜力,为分子科学研究打开了新的图景,并为药物设计、材料科学等领域带来新的可能。

结构预测是分子科学中的一个根本课题,因为分子的三维结构决定了分子的特性和功能。近年来,深度学习方法在分子结构预测方面取得了显著进展,并产生了重大影响。例如,深度学习模型 AlphaFold 和 RoseTTAFold 在从氨基酸序列中预测最有可能的蛋白质结构方面达到了前所未有的准确度;由微软研究院研发的 Graphormer 模型可以精准预测催化剂表面分子的吸附构象,并在全球首届公开催化剂挑战赛中夺冠。尽管深度学习方法改变了分子科学的游戏规则,但为分子的静态结构提供单一快照,仅揭开了复杂分子系统的冰山一角。
以蛋白质分子为例,蛋白质并不是刚性物体,它们是动态的分子,在平衡状态下可以呈现不同的结构,每种结构都有特定的出现概率。平衡分布下的结构及其出现的概率决定了分子的宏观属性和功能,从而才能揭示其生物学原理并对现实应用产生影响。而获得这些平衡分布的传统方法,如分子动力学模拟或蒙特卡洛采样都是从分布中顺序采样,由于其计算成本高,并且采样样本之间统计不独立,所以导致该类方法难以轻易用于复杂的实际应用场景中。因此,分子科学领域迫切需要找到全新方法,可以从分子结构预测问题迈进到分子的平衡分布预测。
DiG:预测平衡态下分子结构的分布
微软研究院发布的全新深度学习框架 Distributional Graphormer (DiG)[1][2],可以用于预测平衡态下分子结构的分布,旨在攻克平衡分布预测这一基础性难题,为分子科学研究创造了新的机遇。DiG 实现了从单一结构预测扩展到对平衡分布的整体预测的重要突破。平衡分布预测弥合了由统计力学和热力学控制的分子系统微观结构和宏观特性之间的差距。这是一项非常具有挑战性的任务,因为它需要对高维空间中的复杂分布进行建模,以捕捉不同分子状态的概率。
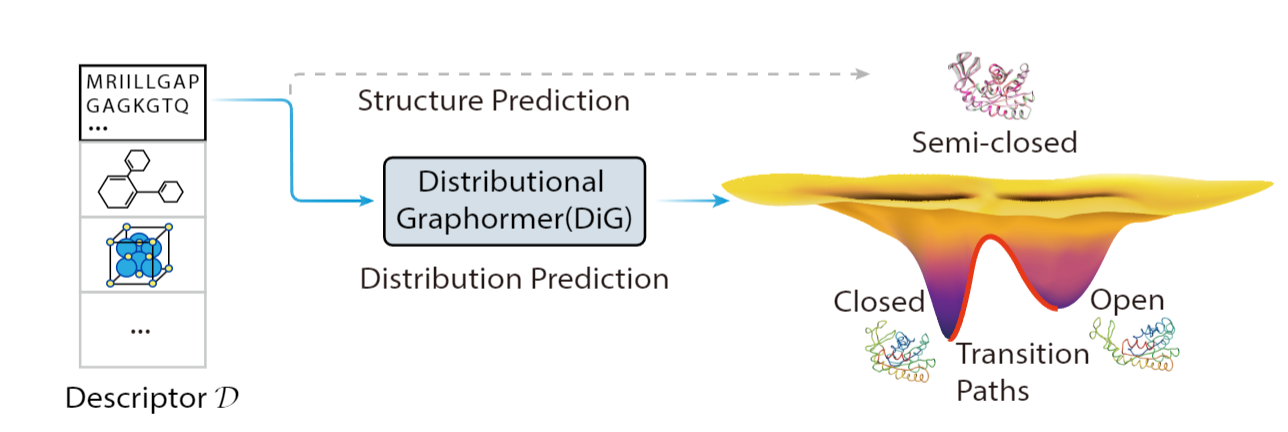
图1:DiG 的目标是以分子系统的基本描述符(例如氨基酸序列或分子化学式)作为输入,预测符合平衡分布的结构及其概率。
通过对此前研究工作 Graphormer 的扩展,DiG 实现了分布预测的全新解决方案。Graphormer 是一种通用的图 (Graph) Transformer,可以有效地对分子结构进行理解和建模,在分子科学中表现出了优异的性能,在量子化学或分子动力学模拟中也得到了应用[3][4]。现在,DiG 具有更新、更强大的功能——通过深度神经网络直接预测平衡分布。
DiG 受到热力学和优化的经典方法——模拟退火算法(simulated annealing)启发,通过模拟一个随机过程,将一个简单分布逐渐完善,从而产生一个复杂分布。此随机过程的预测在深度学习框架中完成。这也是最近将生成式人工智能推向火热的扩散模型(diffusion models)的模式。DiG 将这一思想又带回了热力学研究,形成了一个灵感和创新的闭环。可以想象在不久的未来,科学家们将可以像使用 AI 作画一样来使用 DiG 生成分子结构:通过输入简单的描述,例如氨基酸序列,DiG 就可以快速生成符合平衡分布的、真实多样的分子结构。这将大大提高科学家的生产力和创造力,使其能够在药物设计、材料科学和催化等领域获得新的发现与应用。
在多种分子体系采样任务中,DiG颠覆传统
DiG 框架已在多个分子采样任务上展现出优异的性能和潜力,这些任务涵盖了广泛的分子系统,如蛋白质、蛋白质-配体复合物和催化剂-吸附质系统等。研究结果显示,DiG 不仅能够以高效率和低计算成本生成真实、多样的分子结构,还可以提供状态密度的估计,这对于使用统计力学计算宏观性质至关重要。DiG 在从统计学角度理解微观分子并预测其宏观特性方面取得了重大进展,为分子科学创造了更多令人兴奋的研究机会。
DiG 的重要应用之一是对蛋白质构象进行采样,这对于理解蛋白质性质和功能是必不可少的。蛋白质是动态分子,在平衡状态下会形成不同的结构且形成的概率各不相同,而这些结构通常又与其生物功能和与其他分子的相互作用有关。但是预测蛋白质构象的平衡分布是一个长期存在且具有挑战性的问题,原因在于构象空间中的概率分布取决于复杂和高维的能量景观图(Energy Landscape)。与昂贵且低效的分子动力学模拟或蒙特卡洛采样方法相比,DiG 可以从氨基酸序列中生成多样化并与功能相关的蛋白质结构,不仅速度快,而且成本显著降低。
DiG 可以从相同的蛋白质序列中产生多种构象。如图2所示,DiG 生成了 SARS-CoV-2 病毒主蛋白酶的结构,并与分子动力学模拟和 AlphaFold2 的预测结果进行了比较。在二维空间中,等高线图(以线条表示)显示了由大规模分子动力学模拟采样的三个簇,DiG 在三个簇中均生成了高度相似的结构。

图2:DiG 生成的结构覆盖了新冠病毒主蛋白酶长时间平稳态动力学模拟在2维投影构象空间中分布的主要区域
图3是将 DiG 在四种蛋白质上产生的结构与实验结构进行对比,每种蛋白质都有两种可区分的构象,对应着独特的功能状态。对于左上的腺苷酸激酶蛋白(Adenylate kinase)有开放和闭合状态,两者都被DiG很好地采样。类似地,对于右上的药物转运蛋白 LmrP,DiG 也生成了对应两个功能状态的结构。值得注意的是,闭合状态是通过实验确定的(第二列下方的棕色示例,PDB ID 6t1z),而另一种状态则是与实验数据一致的 AlphaFold2 预测的模型。对于图3左下的人类 B-Raf 激酶而言,主要的结构差异位于 A 环 (A-loop) 区和附近的螺旋,也被 DiG 很好地捕获到了。另一个有趣的例子是具有两个分离结构域的D-核糖结合蛋白(右下),可以被包装成两种不同的构象。虽然 DiG 完美地生成了垂直构象,但未能预测扭曲/倾斜构象。尽管如此,DiG 还是生成了似乎是中间态的构象。总之,DiG 展示了生成与功能相关状态对应的多样化结构的能力,这在此前专注于结构预测的方法中尚未实现。

图3:DiG 在产生蛋白质多种构象方面的性能。在4种不同蛋白,DiG(薄带状)产生的结构与实验确定的结构(圆柱)高度一致。
DiG 的另一个应用是对催化剂-吸附质系统进行采样,这是多相催化的核心。识别活性吸附位点和稳定的吸附质构型是理解和设计催化剂的关键,但由于复杂的表面分子相互作用,这项工作也非常具有挑战性。密度泛函理论(DFT)计算和分子动力学模拟等传统方法往往非常耗时且成本高昂,特别是对于大型的复杂表面。DiG 提供了快速、准确的解决方案,可以根据基质和吸附质描述符,预测吸附位点和构型及其相应的概率。DiG 还可以处理不同类型的吸附质,如单原子或分子,以及金属或合金等不同类型的基质。
通过 DiG,研究员们预测了各种催化剂-吸附质系统的吸附位点,并将预测结果与 DFT 计算得到的能量进行了比较。如图4所示,DiG 可以找到所有稳定的吸附位点,并产生类似于 DFT 结果的吸附质构型,效率高且成本低。DiG 还可以估算不同吸附构型的形成概率,这与 DFT 能量非常一致。

图4:单个 N 原子和 O 原子在催化剂表面的吸附预测结果。模型预测的催化剂表面吸附质吸附概率分布与量子化学计算得到的相互作用能分部对比图。
DiG还在蛋白-配体采样,逆设计等任务中展现了前所未有的能力。具体内容请参考论文原文。
DiG是如何工作的?
类似于模拟退火过程的模式,DiG 通过使用 Graphormer 模型预测一个扩散过程,将简单分布转换为复杂分布。简单分布通常是标准高斯分布,复杂分布则是分子结构的平衡分布。转换是一步一步进行的,如此建模复杂分布的难度便被拆解到每一步成为较为简单的问题。

图5:DiG 的设计和骨干架构
DiG 可以使用不同类型的数据或信息来进行训练。DiG 首先可以使用模拟数据,例如分子动力学轨迹,来学习分布。DiG 也可以直接使用分子系统的能量函数来训练,因为平衡分布可通过统计力学理论直接由能量函数给出。由于分子体系平衡分布预测不同于传统 AI 任务,其数据生成需要耗费长时间的模拟计算因而难以大规模得到,直接从能量函数学习便是一个缓解对数据严格依赖的手段。
DiG 在许多分子系统上都显示出与基于深度学习的结构预测方法相似的良好泛化能力。这是因为 DiG 继承了先进的深度学习架构,如 Graphormer 的优势,并将其应用于一个新的、具有挑战性的分布预测任务。训练好后,DiG 可以通过反转转换过程来生成分子结构,从一个简单的分布开始,并以相反的顺序调用深度学习模型。DiG 还可以通过计算转换过程中概率的变化来提供每个生成结构的概率估计。可以看到,DiG 是一个灵活而通用的框架,可以处理不同类型的分子系统和描述符。
未来,为分子科学研究开辟更多新机遇
DiG 是从单一结构预测到对平衡分布整体建模的重大进展,为在深度学习框架下连接微观结构和宏观属性奠定了基石。DiG 使用生成式 AI 技术,可以在多种分子系统中对符合平衡分布的分子结构进行采样。研究员们在包括蛋白质在内的不同类别的分子上展示了 DiG 的灵活性,同时也证明了以这种方式生成的单一结构是符合物理化学相互作用规律的。
然而,要获得对任意分子系统平衡分布更精准的预测,仍需要进行更多的研究。微软研究院希望 DiG 能够沿着这一方向激发更多的研究与创新,期待未来能够看到 DiG 和其他方法在分子平衡分布预测问题上带来更多令人兴奋的成果和影响。
相关链接:
[1] DiG 论文:Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning (https://www.microsoft.com/en-us/research/publication/towards-predicting-equilibrium-distributions-for-molecular-systems-with-deep-learning/)
[2] Demo页面 (https://distributionalgraphormer.github.io)
[1] KDD Cup 2021 | 微软亚洲研究院Graphormer模型荣登OGB-LSC图预测赛道榜首 (https://www.msra.cn/zh-cn/news/features/ogb-lsc)
[2] 公开催化剂挑战赛冠军模型、通用AI分子模拟库Graphormer开源!(https://www.msra.cn/zh-cn/news/features/graphormer)