笔记来自《药物设计学》
文章目录
- 1.药物设计的分子基础
- 2.药物设计的理论基础
1.药物设计的分子基础
- 化学小分子药物与生物大分子靶标之间相互作用是药物设计的分子基础
- 临床所用的药物50%都属于手性化合物,这是因为药物靶标的基本单元如氨基酸或核苷酸都是手性结构
- 区分构型(configuration)和构象(conformation):
- 构型是分子结构相同,但原子的空间排列不同,包括顺反异构,立体异构等
- 构象是构型相同时由于单键旋转造成的不同构象异构
- 手性是分子构型的一个重要因素,利用偏振光区分相对构型:左旋是(-)或 l,右旋是(+)或 d,利用空间排列区分绝对构型:R/S构型
- 对映异构体的生物学效应可能表现出强弱、相反、不相关、治疗和毒性,互补等关系
- 辉瑞研究员Lipinski根据2245个药物总结出成药性“五规则”(近年来又有陆续补充的经验性规则):
- 分子量不超过500
- 亲脂性clogp不超过5
- 氢键供体数不超过5
- 氢键受体数不超过10
- 药物分子可分为侧链部分(side chain)和骨架(scaffold 或 framework)部分
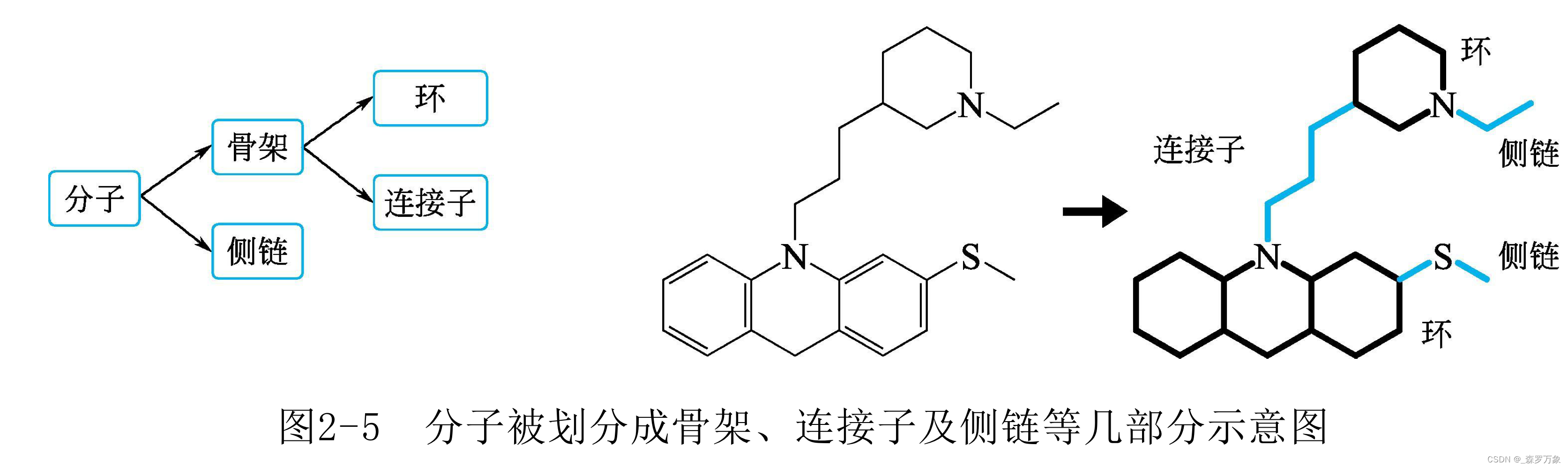
- 常用的类药学骨架是优势结构,公认的毒性基团或化学反应性基团是警示子结构(structural alerts)
- 先导化合物的优化过程中,应该尽量减少先导化合物中的冗余原子,配体效率(Ligand Efficiency)是每个重原子对结合能的贡献均值 L A = Δ G H A LA=\frac{\Delta G}{HA} LA=HAΔG,HA 是重原子数目。较高的配体效率可能使得药代动力学性质提高
- 肽键中存在两个二面角,这两个二面角决定了多肽链的走向。由于原子间的立体碰撞,许多角度组合不被允许,以两个二面角的角度画图即为拉式图,可以用于判断蛋白质结构是否合理,用于同源建模
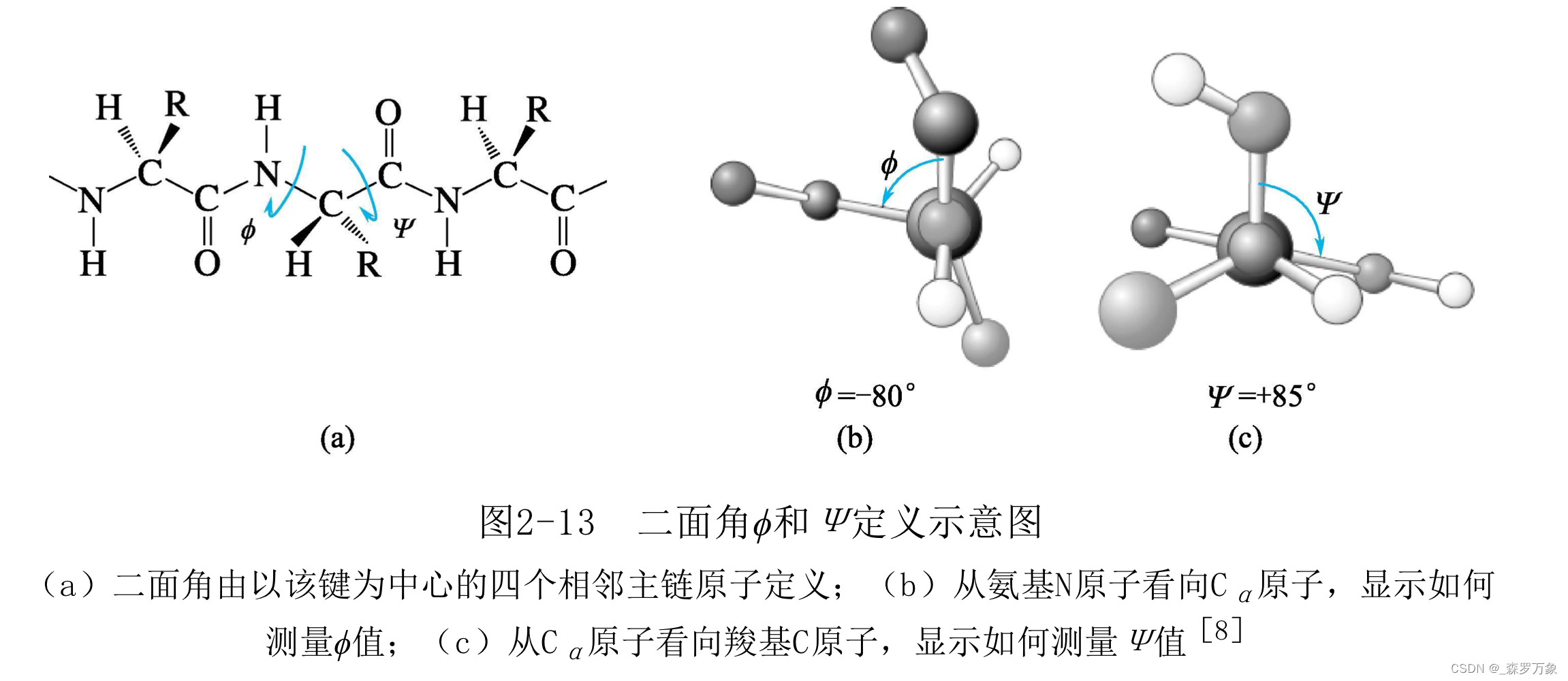
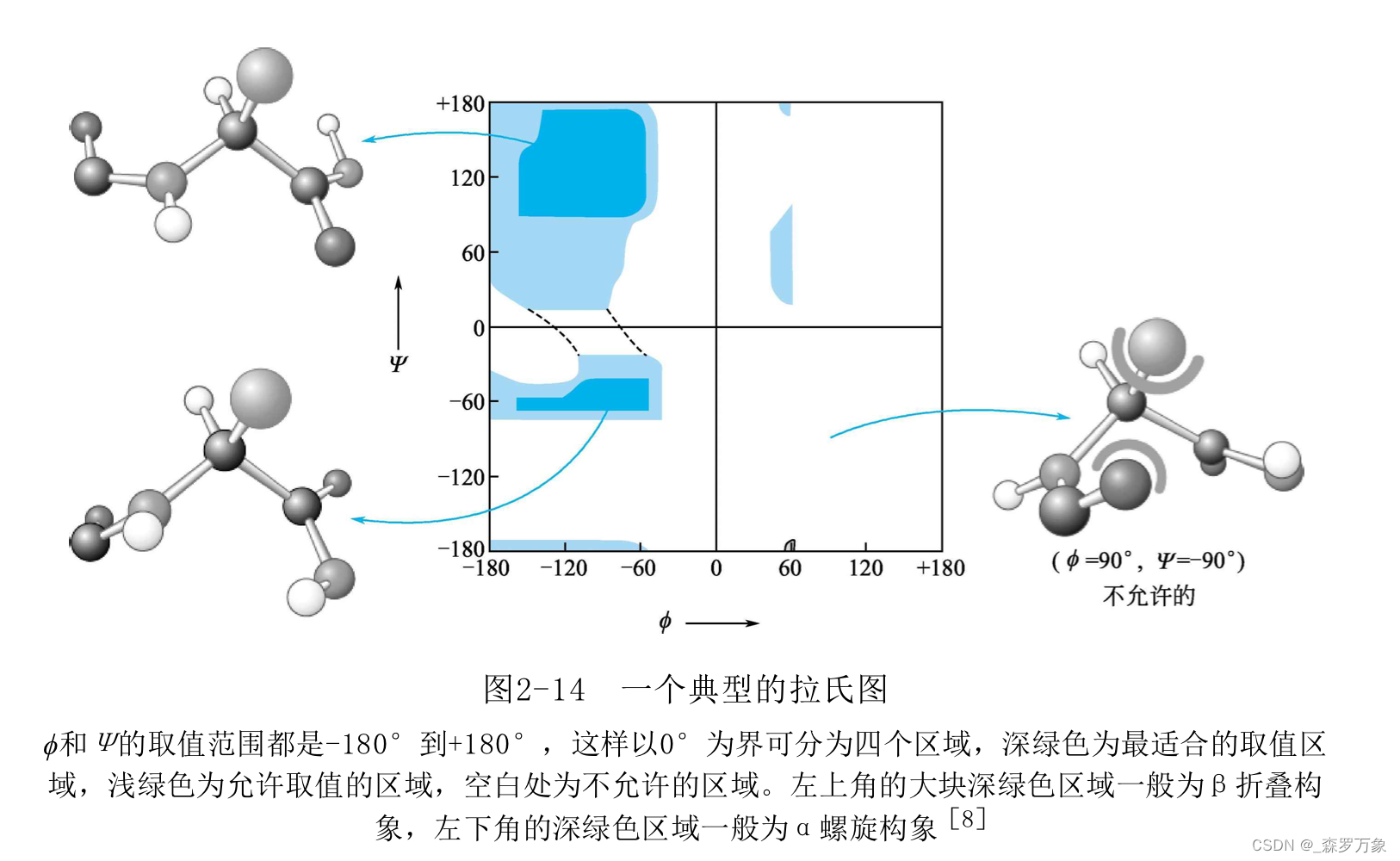
- 酶与底物结合的位点是活性位点,与配体结合的口袋是变构位点,变构位点结合后可以改变酶的活性构象
- 酶分为单纯酶和结合酶两大类,后者与辅酶因子结合后是全酶,只有全酶才有催化功能。酶一般以-ase为后缀
- 受体分为G蛋白(鸟苷酸结合蛋白)偶联受体,离子通道受体,单跨膜受体(如酪氨酸激酶受体),核受体
- 多糖也有四级结构,其一级结构比蛋白质和核酸复杂得多
- RNA分子同蛋白质分子一样具有结构多样性和明确的配体结合腔体
- 药物与靶标的结合是“诱导-契合”,药物结合靶标之后,靶标会逐渐适应药物,出现性质互补和性质互补
- 生物分子之间的相互作用包括亲和力(binding affinity)和专一性(specificity),前者是分子之间的结合,后者是结合之后产生生物学效应。