欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/131461900
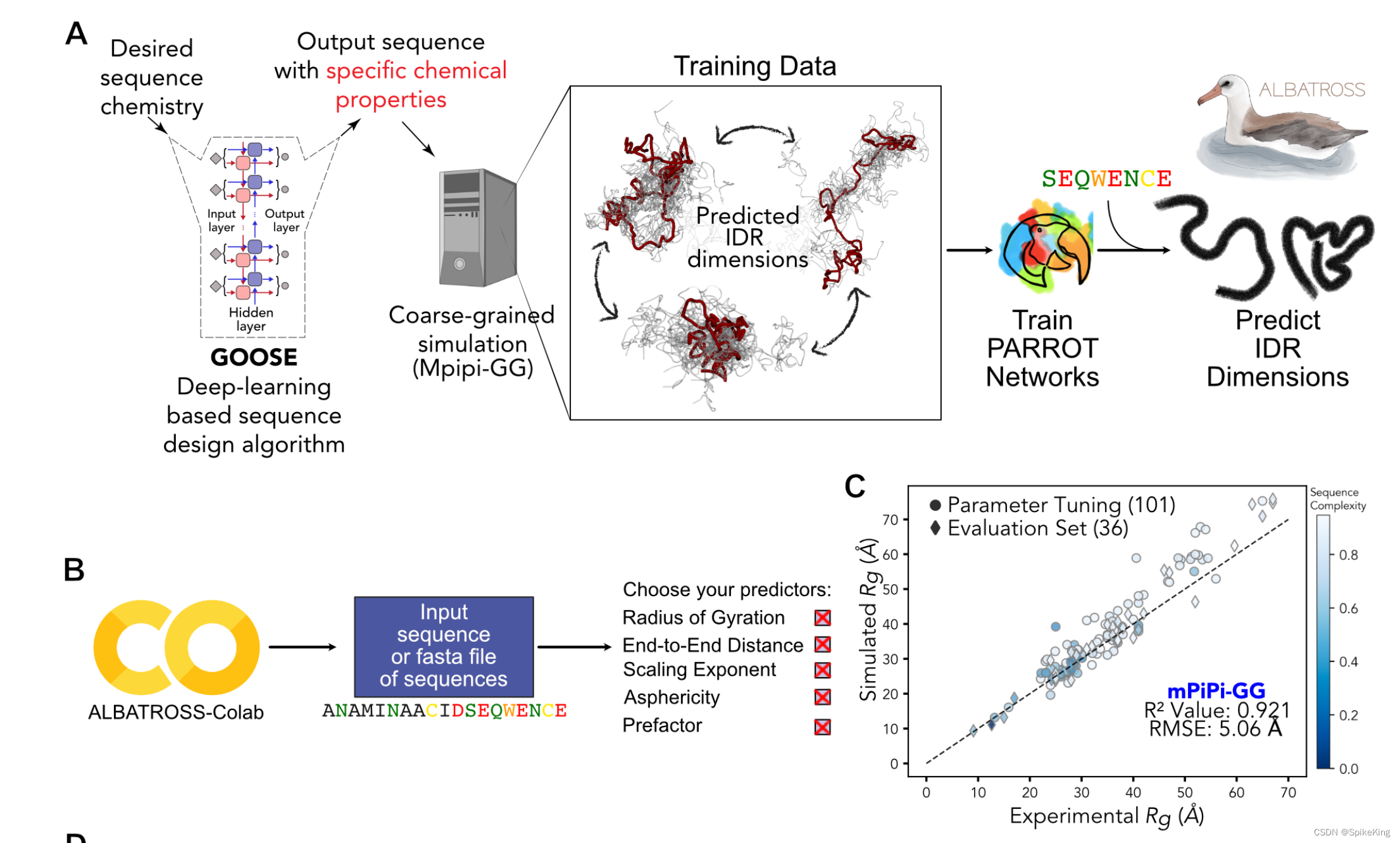
MetaPredict 算法简介:
内源性无序区域(IDRs)在所有生命领域中都普遍存在,并发挥着各种功能作用。虽然折叠结构域通常可以用一个三维结构来很好地描述,但是 IDRs 存在于一系列相互转化的状态中,称为集合体。这种结构异质性意味着 IDRs 在 PDB 中大部分缺失,导致缺乏从序列预测集合体构象特性的计算方法。在这里,我们结合了合理的序列设计、大规模的分子模拟和深度学习,开发 ALBATROSS,一个用于从序列预测 IDR 集合体尺寸的深度学习模型。ALBATROSS 能够瞬间预测蛋白质组范围内的集合体平均特性。ALBATROSS 轻量、易用,既可以作为一个本地安装的软件包,也可以作为一个点击式的云端界面。我们首先通过检验IDRs中序列-集合体关系的泛化性,来展示我们的预测器的适用性。然后,我们利用 ALBATROSS 的高通量特性,来表征 IDRs 在蛋白质组内外的新出现的生物物理行为。
使用工具 metapredict:
-
GitHub:metapredict: A machine learning-based tool for predicting protein disorder.
-
使用文档:https://metapredict.readthedocs.io/en/latest/
Paper:Direct prediction of intrinsically disordered protein conformational properties from sequence
- 更新时间 2023-05-28
在线接口:https://metapredict.net/
参考的测试文档,来自 @盼盼: https://wiki.biomap-int.com/pages/viewpage.action?pageId=100631778
1. 工程配置
测试 T1157s1_A1029.fasta,来自于 CASP15 :
>A
DRVRALRRETVEMFYYGFDNYMKVAFPEDELRPVSCTPLTRDLKNPRNFELNDVLGNYSLTLIDSLSTLAILASAPAEDSGTGPKALRDFQDGVAALVEQYGDGRPGPSGVGRRARGFDLDSKVQVFETVIRGVGGLLSAHLFAIGALPITGYQPLRQEDDLFNPPPIPWPNGFTYDGQLLRLALDLAQRLLPAFYTKTGLPYPRVNLRHGIPFYVNSPLHEDPPAKGTTEGPPEITETCSAGAGSLVLEFTVLSRLTGDPRFEQAAKRAFWAVWYRKSQIGLIGAGVDAEQGHWIGTYSVIGAGADSFFEYALKSHILLSGHALPNQTHPSPLHKDVNWMDPNTLFEPLSDAENSAESFLEAWHHAHAAIKRHLYSEREHPHYDNVNLWTGSLVSHWVDSLGAYYSGLLVLAGEVDEAIETNLLYAAIWTRYAALPERWSLREKTVEGGLGWWPLRPEFIESTYHLYRATKDPWYLYVGEMVLRDITRRCWTPCGWAGLQNVLSGEKSDRMESFFLGETTKYMYLLFDDDHPLNKLDASFVFTTEGHPLILPKPKSARRSRNSPRSSQKALTVYQGEGFTNSCPPRPSITPLSGSVIAARDDIYHPARMVDLHLLTTSKHALDGGQMSGQHMAKSNYTLYPWTLPPELLPSNGTCAKVYQPHEVTLEFASNTQQVLGGSAFNFMLSGQNLERLSTDRIRVLSLSGLKITLQLVEEGEREWRVTKLNGIPLGRDEYVVINRAILGDVSDPRFNLVRDPVIAKLQQLHQVNLLDDTTTEEHPDNLDTLDTASAIDLPQDQSSDSEVPDPANLSALLPDLSSFVKSLFARLSNLTSPSPDPSSNLPLNVVINQTAILPTGIGAAPLPPAASNSPSGAPIPVFGPVPESLFPWKTIYAAGEACAGPLPDSAPRENQVILIRRGGCSFSDKLANIPAFTPSEESLQLVVVVSDDEHEGQSGLVRPLLDEIQHTPGGMPRRHPIAMVMVGGGETVYQQLSVASAIGIQRRYYIESSGVKVKNIIVDDGDGGVDG
安装 Python 包:
pip install metapredict==2.61
import metapredict as meta
2. 函数调用
2.1 核心函数 Predict Disorder Batch
测试,计算 Residue Disorder 的概率值0~1,值越大表示越可能是 Disorder 位点,阈值取 0.5,映射成0、1二值化,1 表示 disorder,0 表示 fold。
def predict_batch(seq_list):
if not isinstance(seq_list, list):
seq_list = [seq_list]
output = meta.predict_disorder_batch(seq_list)
assert len(seq_list) == len(output)
res_list = []
for sample in output:
sample_disorder = sample[1]
print(f"disorder range: {np.min(sample_disorder)}~{np.max(sample_disorder)}")
sample_disorder_idx = list(np.where(sample_disorder > 0.5, 1, 0))
print(f"sample_disorder_idx: {sample_disorder_idx}")
# 获取 disorder 区间
d_list, tmp_list = [], []
for i, v in enumerate(sample_disorder_idx):
if v == 1: # 无序
tmp_list.append(i)
else:
if tmp_list:
d_list.append(copy.copy(tmp_list))
tmp_list = []
domain = []
for r in d_list:
if (r[-1] - r[0]) >= 2:
domain.append([r[0], r[-1]])
# seq, disorder_idx, domain
res_list.append([sample[0], sample_disorder_idx, domain])
return res_list
其中,阈值 0.5,来自于算法建议值。
Altering the disorder theshhold - To alter the disorder threshold, simply set
disorder_threshold=my_valuewheremy_valueis a float. The higher the threshold value, the more conservative metapredict will be for designating a region as disordered. Default = 0.5 (V2) and 0.42 (legacy).
输出:
disorder range: 0.0~0.9868000149726868
seq:
DRVRALRRETVEMFYYGFDNYMKVAFPEDELRPVSCTPLTRDLKNPRNFELNDVLGNYSLTLIDSLSTLAILASAPAEDSGTGPKALRDFQDGVAALVEQYGDGRPGPSGVGRRARGFDLDSKVQVFETVIRGVGGLLSAHLFAIGALPITGYQPLRQEDDLFNPPPIPWPNGFTYDGQLLRLALDLAQRLLPAFYTKTGLPYPRVNLRHGIPFYVNSPLHEDPPAKGTTEGPPEITETCSAGAGSLVLEFTVLSRLTGDPRFEQAAKRAFWAVWYRKSQIGLIGAGVDAEQGHWIGTYSVIGAGADSFFEYALKSHILLSGHALPNQTHPSPLHKDVNWMDPNTLFEPLSDAENSAESFLEAWHHAHAAIKRHLYSEREHPHYDNVNLWTGSLVSHWVDSLGAYYSGLLVLAGEVDEAIETNLLYAAIWTRYAALPERWSLREKTVEGGLGWWPLRPEFIESTYHLYRATKDPWYLYVGEMVLRDITRRCWTPCGWAGLQNVLSGEKSDRMESFFLGETTKYMYLLFDDDHPLNKLDASFVFTTEGHPLILPKPKSARRSRNSPRSSQKALTVYQGEGFTNSCPPRPSITPLSGSVIAARDDIYHPARMVDLHLLTTSKHALDGGQMSGQHMAKSNYTLYPWTLPPELLPSNGTCAKVYQPHEVTLEFASNTQQVLGGSAFNFMLSGQNLERLSTDRIRVLSLSGLKITLQLVEEGEREWRVTKLNGIPLGRDEYVVINRAILGDVSDPRFNLVRDPVIAKLQQLHQVNLLDDTTTEEHPDNLDTLDTASAIDLPQDQSSDSEVPDPANLSALLPDLSSFVKSLFARLSNLTSPSPDPSSNLPLNVVINQTAILPTGIGAAPLPPAASNSPSGAPIPVFGPVPESLFPWKTIYAAGEACAGPLPDSAPRENQVILIRRGGCSFSDKLANIPAFTPSEESLQLVVVVSDDEHEGQSGLVRPLLDEIQHTPGGMPRRHPIAMVMVGGGETVYQQLSVASAIGIQRRYYIESSGVKVKNIIVDDGDGGVDG
disorder list:
[1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 1, 1, 1]
disordered domains:
[[224, 230], [553, 570], [772, 810], [832, 880]]
2.2 Disorder Domains (Official)
predict_disorder_domains 的 disorder 预测值与 predict_disorder_batch 一致,只是选择 domains 的条件不同,范围更大。
源码:
output = meta.predict_disorder_domains(seq)
print(output)
print(f"disorder: {output.disorder}")
print(f"disordered_domain_boundaries: {output.disordered_domain_boundaries}")
for boundary in output.disordered_domain_boundaries:
s = boundary[0]
e = boundary[1]
print(f"disorder: {np.min(output.disorder[s:e])} ~ {np.max(output.disorder[s:e])}")
print(f"folded_domain_boundaries: {output.folded_domain_boundaries}")
print(f"disordered_domains: {output.disordered_domains}")
print(f"folded_domains: {output.folded_domains}")
输出:
DisorderObject for sequence with 1029 residues, 2 IDRs, and 3 folded domains
Available dot variables are:
.sequence
.disorder
.disordered_domain_boundaries
.folded_domain_boundaries
.disordered_domains
.folded_domains
disorder: [0.5593 0.5207 0.4646 ... 0.7331 0.7067 0.6378]
disordered_domain_boundaries: [[553, 571], [772, 880]]
disorder: 0.5241 ~ 0.854
disorder: 0.2163 ~ 0.9868
folded_domain_boundaries: [[0, 553], [571, 772], [880, 1029]]
disordered_domains: ['KPKSARRSRNSPRSSQKA', 'DDTTTEEHPDNLDTLDTASAIDLPQDQSSDSEVPDPANLSALLPDLSSFVKSLFARLSNLTSPSPDPSSNLPLNVVINQTAILPTGIGAAPLPPAASNSPSGAPIPVF']
folded_domains: ['DRVRALRRETVEMFYYGFDNYMKVAFPEDELRPVSCTPLTRDLKNPRNFELNDVLGNYSLTLIDSLSTLAILASAPAEDSGTGPKALRDFQDGVAALVEQYGDGRPGPSGVGRRARGFDLDSKVQVFETVIRGVGGLLSAHLFAIGALPITGYQPLRQEDDLFNPPPIPWPNGFTYDGQLLRLALDLAQRLLPAFYTKTGLPYPRVNLRHGIPFYVNSPLHEDPPAKGTTEGPPEITETCSAGAGSLVLEFTVLSRLTGDPRFEQAAKRAFWAVWYRKSQIGLIGAGVDAEQGHWIGTYSVIGAGADSFFEYALKSHILLSGHALPNQTHPSPLHKDVNWMDPNTLFEPLSDAENSAESFLEAWHHAHAAIKRHLYSEREHPHYDNVNLWTGSLVSHWVDSLGAYYSGLLVLAGEVDEAIETNLLYAAIWTRYAALPERWSLREKTVEGGLGWWPLRPEFIESTYHLYRATKDPWYLYVGEMVLRDITRRCWTPCGWAGLQNVLSGEKSDRMESFFLGETTKYMYLLFDDDHPLNKLDASFVFTTEGHPLILP', 'LTVYQGEGFTNSCPPRPSITPLSGSVIAARDDIYHPARMVDLHLLTTSKHALDGGQMSGQHMAKSNYTLYPWTLPPELLPSNGTCAKVYQPHEVTLEFASNTQQVLGGSAFNFMLSGQNLERLSTDRIRVLSLSGLKITLQLVEEGEREWRVTKLNGIPLGRDEYVVINRAILGDVSDPRFNLVRDPVIAKLQQLHQVNLL', 'GPVPESLFPWKTIYAAGEACAGPLPDSAPRENQVILIRRGGCSFSDKLANIPAFTPSEESLQLVVVVSDDEHEGQSGLVRPLLDEIQHTPGGMPRRHPIAMVMVGGGETVYQQLSVASAIGIQRRYYIESSGVKVKNIIVDDGDGGVDG']
2.3 Predict pLDDT
metapredict 支持预测 Residue 的 pLDDT 值,用于评估序列的质量。
源码:
output = meta.predict_pLDDT(seq)
print(f"mean_plddt: {np.mean(output)}")
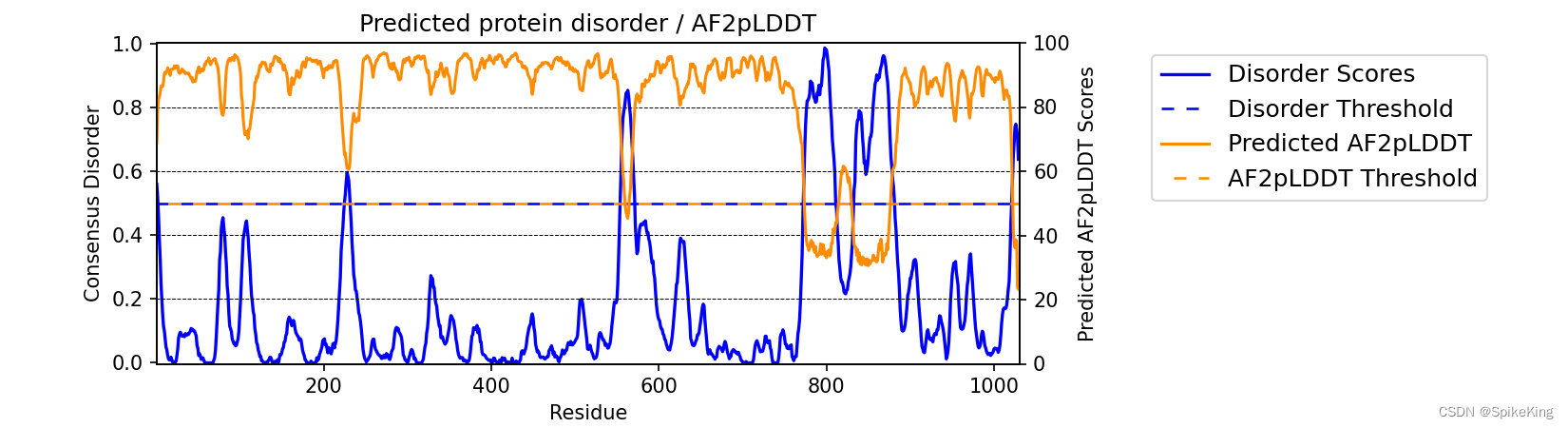
2.4 绘图 Graph Disorder
直接调用 graph_disorder,即可绘图:
meta.graph_disorder(seq, pLDDT_scores=True)
Disorder Scores 与 pLDDT 成负相关,绘图如下:
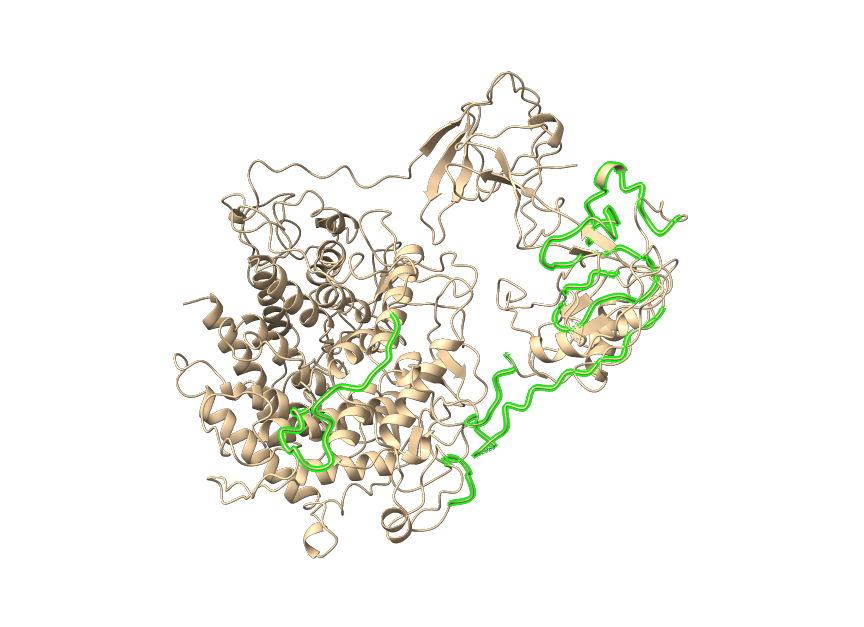
3. 测试结构
使用 ChimeraX 测试结构,命令脚本如下:
@classmethod
def get_chimerax_select_cmd(cls, seq_list, mod_num="1"):
"""
[kaɪˈmɪrə]
select #1:553-571 #1:772-880
"""
res_list = cls.predict_batch(seq_list)
r_str_list = []
for res in res_list:
domains = res[2]
c_list = ["select"]
for domain in domains:
c_str = f"#{mod_num}:{domain[0]}-{domain[1]}"
c_list.append(c_str)
r_str = " ".join(c_list)
r_str_list.append(r_str)
return r_str_list
输出:
ChimeraX: select #1:224-230 #1:553-570 #1:772-810 #1:832-880
测试结构,绿色高亮就是无序区:
其他
参考:
-
Paper - Direct Prediction of Intrinsically Disordered Protein Conformational Properties From Sequence
-
metapredict online (v2.3)
-
chimerax-example-scripts-commands
关于 ChatGPT 的翻译 Prompt:
您好,如果你准备好了,请回答“是”,以下是一段蛋白质相关的段落,你作为一个生物学家,帮忙翻译成中文。"[需要翻译的段落]"
源码如下:
#!/usr/bin/env python
# -- coding: utf-8 --
"""
Copyright (c) 2022. All rights reserved.
Created by C. L. Wang on 2023/6/29
"""
import copy
import os
import metapredict as meta
import numpy as np
from protein_utils.seq_utils import get_seq_from_fasta
from root_dir import DATA_DIR
class SeqIdrPredictor(object):
"""
序列的 IDRs 区域预测
pip install metapredict==2.61
"""
def __init__(self):
pass
@staticmethod
def predict_batch(seq_list):
"""
核心函数
"""
if not isinstance(seq_list, list):
seq_list = [seq_list]
output = meta.predict_disorder_batch(seq_list)
assert len(seq_list) == len(output)
res_list = []
for sample in output:
sample_disorder = sample[1]
print(f"disorder range: {np.min(sample_disorder)}~{np.max(sample_disorder)}")
sample_disorder_idx = list(np.where(sample_disorder > 0.5, 1, 0))
# 获取 disorder 区间
d_list, tmp_list = [], []
for i, v in enumerate(sample_disorder_idx):
if v == 1: # 无序
tmp_list.append(i)
else:
if tmp_list:
d_list.append(copy.copy(tmp_list))
tmp_list = []
domains = []
for r in d_list:
if (r[-1] - r[0]) >= 2:
domains.append([r[0], r[-1]])
# seq, disorder_idx, domain
res_list.append([sample[0], sample_disorder_idx, domains])
return res_list
@classmethod
def get_chimerax_select_cmd(cls, seq_list, mod_num="1"):
"""
[kaɪˈmɪrə]
select #1:553-571 #1:772-880
"""
res_list = cls.predict_batch(seq_list)
r_str_list = []
for res in res_list:
domains = res[2]
c_list = ["select"]
for domain in domains:
c_str = f"#{mod_num}:{domain[0]}-{domain[1]}"
c_list.append(c_str)
r_str = " ".join(c_list)
r_str_list.append(r_str)
return r_str_list
@staticmethod
def predict_disorder_domains(seq, is_print=False):
output = meta.predict_disorder_domains(seq)
if is_print:
output = meta.predict_disorder_domains(seq)
print(output)
print(f"disorder: {output.disorder}")
print(f"disordered_domain_boundaries: {output.disordered_domain_boundaries}")
for boundary in output.disordered_domain_boundaries:
s = boundary[0]
e = boundary[1]
print(f"disorder: {np.min(output.disorder[s:e])} ~ {np.max(output.disorder[s:e])}")
print(f"folded_domain_boundaries: {output.folded_domain_boundaries}")
print(f"disordered_domains: {output.disordered_domains}")
print(f"folded_domains: {output.folded_domains}")
return output
def main():
fasta_dir = os.path.join(DATA_DIR, "CASP15-Monomer-Targets-56", "fasta")
fasta_path = os.path.join(fasta_dir, "T1157s1_A1029.fasta")
seq = get_seq_from_fasta(fasta_path)[0]
sip = SeqIdrPredictor()
res_list = sip.predict_batch(seq)
print(f"seq:\n{res_list[0][0]}")
print(f"disorder list:\n{res_list[0][1]}")
print(f"disordered domains:\n{res_list[0][2]}")
sip.predict_disorder_domains(seq, is_print=True)
output = meta.predict_pLDDT(seq)
print(f"mean_plddt: {np.mean(output)}")
meta.graph_disorder(seq, pLDDT_scores=True)
r_str_list = sip.get_chimerax_select_cmd(seq)
print(f"ChimeraX: {r_str_list[0]}")
if __name__ == '__main__':
main()