在解析x射线蛋白晶体结构过程中,最常用的方法就是分子置换,即进行molecular replacement(MR)时,输入合适的同源蛋白作为model,以及目标蛋白的mtz和sequence,来完成相位解析的过程。
相位求解成功与否除了查看MR结束后的TFZ score(通常应该至少大于10)和LLG value(通常应该至少大于200)之外,还可通过后续的refmac查看Rfree(最好小于0.4)是否正常。
相位求解不成功的因素有很多,比如数据本身的质量问题,或者是空间群晶胞参数等设置问题。还有一个常见的问题就是model的选择问题,可以尝试着换一个model。
本文想介绍一下如果MR解正常,但是Rfree很高的情况下,如何利用CCP4i2中的Autobuild来创建一个新的model解决的方法。但是这个方法也并不绝对。可能有时候管用有时候不管用。
终端输入ccp4i2打开界面:
$ccp4i2
根据提示新建好project后,选择Autobuild protein
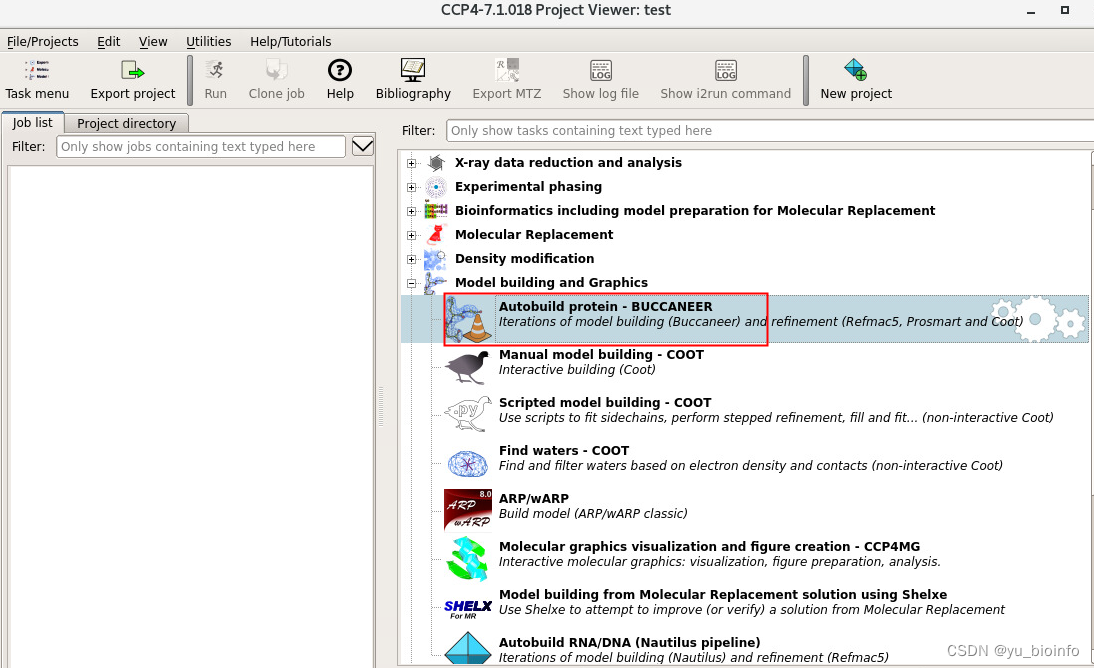
根据提示依次输入文件
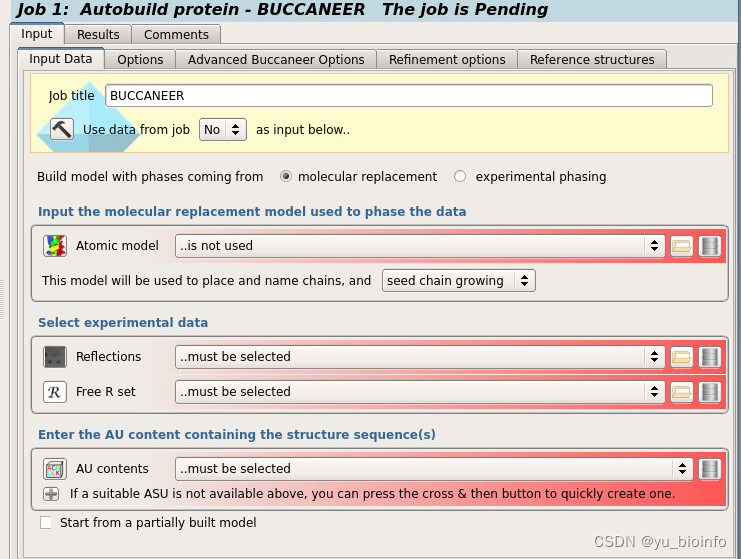
Atomic model:输入上一步MR后的pdb。
Reflections and Free R set: 均为上一步MR后的对应mtz
这里解释一下为什么是MR后的pdb和mtz,因为用已知的model去解析时,MR结果正常,但是RFree数值不理想,所以解是对的,然后尝试用这个方法去重新在此基础上创建一个新的model,再重复做一次MR,看能否降低Rfree。
AU contents: 溶剂含量这里不支持直接输入,需要自动重新算一次。
Start from a partially built model:这里前面的小方框可以勾选上,然后输入和Atomic model一样的pdb文件。
点击上方Run开始运行,以及结果查看
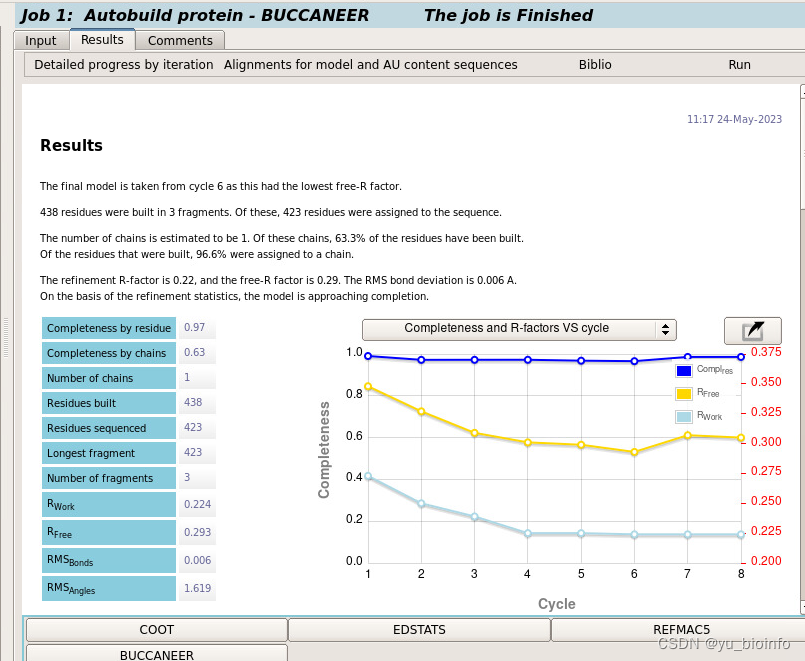
结果model各项指标正常。可用它当model再重新做一次MR。