原文标题:Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
代码:https://github.com/aspuru-guzik-group/chemical_vae
原文链接:https://pubs.acs.org/doi/10.1021/acscentsci.7b00572
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
一、Autoencoder Architecture.
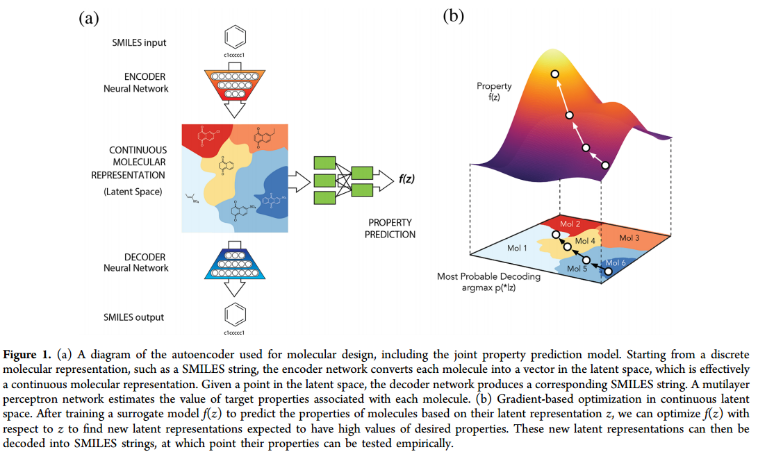
编码器RNN可以与解码器RNN配对以执行序列到序列的学习,还用卷积网络进行了字符串编码的实验,并观察到性能的提高。这可以通过重复的、平移不变的子串的存在来解释,这些子串对应于化学子结构,例如环和官能团。
基于smiles的文本编码对ZINC和QM9分别使用了35个不同字符和22个不同字符的子集。为了便于计算,将字符串编码为ZINC的最大长度为120个字符,QM9的最大长度为34个字符。较短的字符串用相同长度的空格填充。只使用规范化的SMILES进行训练,以避免处理等效的SMILES表示。
VAE深度网络的结构如下: 对于用于ZINC数据集的自编码器,编码器分别使用滤波器大小为9,9,10和9,9,11卷积核的三个1D卷积层,后面是一个宽度为196的全连接层。解码器输入到三层门控循环单元(GRU)网络中,隐藏维数为488。
对于QM9数据集所使用的模型,编码器使用了3个一维卷积层,滤波器大小分别为2,2,1和5,5,4个卷积核,然后是一个宽度为156的全连接层。三个递归神经网络层各有500个神经元的隐藏维度。
潜在空间中的同一点可能解码成不同的SMILES字符串,这取决于用于采样字符的随机种子。输出GRU层有一个额外的输入,对应于从前一个时间步的softmax输出中采样的字符,并使用teacher forcing进行训练,增加了生成的SMILES字符串的准确性,这导致训练数据之外的潜在点的有效SMILES字符串的比例更高,但也使训练变得更加困难,因为解码器显示出忽略(变分)编码并完全依赖输入序列的倾向。29个epoch后,按sigmoid schedule退火,共运行120个epoch
对于属性预测,使用两个全连接(1000个神经元)层来预测潜在表示的属性,dropout为0.20。为了简单地塑造潜在空间,使用了一个较小的MLP作为属性预测器,该MLP由3层67个神经元组成,训练dropout为0.15。对于在ZINC数据集上训练的算法,目标属性包括logP、QED、SAS。性能预测损失与变分损失同时退火。TensorFlow开发
二、RESULTS AND DISCUSSION
1、Representation of Molecules in Latent Space.
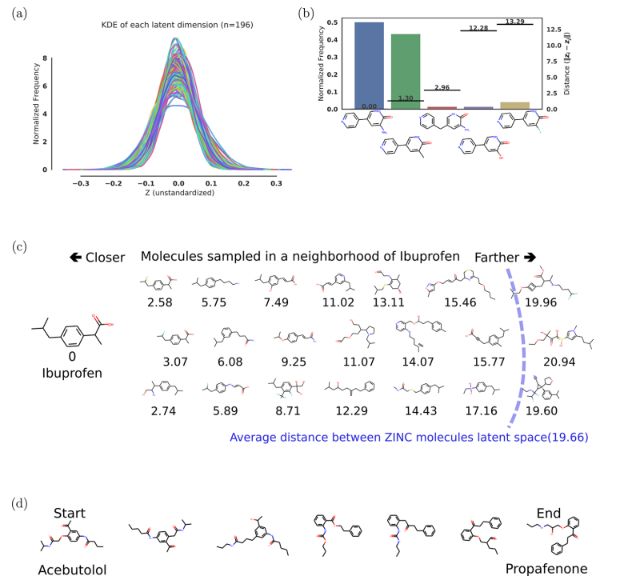
图2a显示了从训练集外随机选择的5000个ZINC分子编码时每个维度的核密度估计。核密度估计显示了数据点沿潜在空间各维的分布。虽然数据点在每个单独维度上的分布显示出略有不同的平均值和标准差,但由于变分正则化器,所有分布都是正态分布。
图2b显示了将fda批准的药物分子样本的潜在表征解码为几个不同分子的概率。对于大多数潜在点,一个突出的分子被解码,许多其他轻微的变化以较低的频率出现。当这些生成的SMILES被重新编码到潜在空间时,最常见的解码也往往是与原始点欧几里得距离最小的那个,这表明潜在空间确实捕获了与分子相关的特征。
图2c显示了潜伏空间中接近布洛芬的一些分子。这些结构变得不那么类似于增加潜在空间的距离。当距离接近训练集中分子的平均距离时,变化更加明显,最终类似于可能从训练集中采样的随机分子。
图2d显示了两个随机药物分子之间的球面插值,显示了两者之间的平滑过渡。连续的潜空间允许分子内插,通过在它们的潜表示之间遵循最短的欧几里得路径。在探索高维空间时,重要的是要注意欧几里得距离可能不能直接映射到分子相似性的概念在高维空间中,大多数独立的正态分布随机变量的质量不是在平均值附近,而是在平均值周围的一个环中两点之间的线性插值可能会经过一个低概率区域,为了使采样保持在高概率区域,使用球面插值35 (slerp)。对于slerp,两点之间的路径是位于n维球体表面上的圆弧。
表1将训练集中的化学性质分布与遗传算法生成的分子和变分自编码器生成的分子进行比较。该过程使用锌数据集中的1000个随机分子进行播种,并在10次迭代中生成。对于使用变分自编码器生成的分子,我们收集了从相同的1000个种子分子编码的潜在空间点进行400次解码尝试生成的所有分子的集合。比较logP、SA和QED。
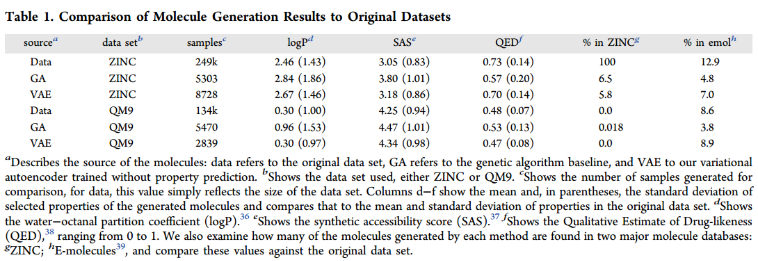
与遗传算法生成的分子集相比,使用VAE生成的分子显示出与原始数据集更相似的化学性质。
2、Property Prediction of Molecules.
训练了MLP和AE来预测每个分子的潜在表征,图3显示了属性值到潜在值的映射。PCA降维。值高的分子位于一个区域,值低的分子位于另一个区域。
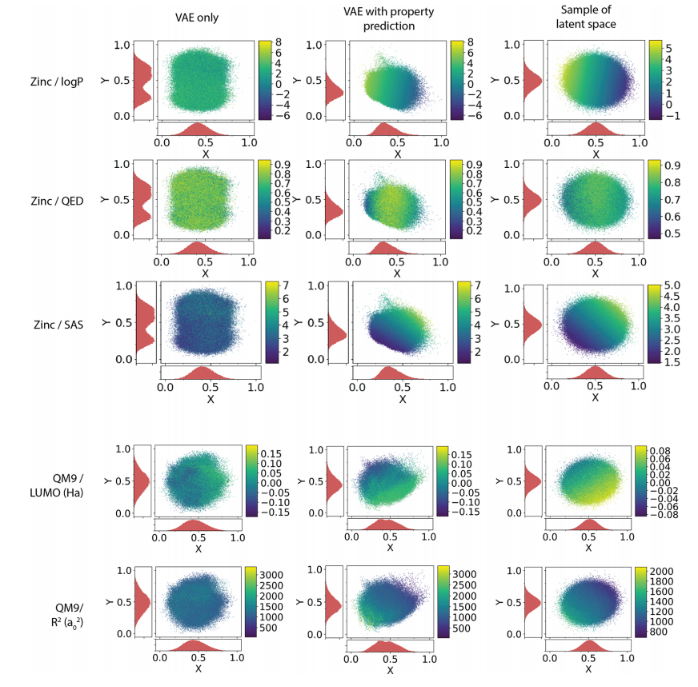
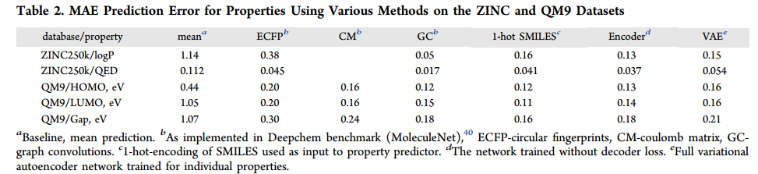
虽然添加性质预测的主要目的是组织潜在空间,但观察属性预测器模型与其他属性预测标准模型的比较是很有趣的。为了与其他方法进行更公平的比较,我们将感知器的大小增加到两层,每层有1000个神经元。表2比较了常用的分子embedding和模型与VAE的性能。
3、Optimization of Molecules via Properties.
从联合训练的自编码器中对潜在空间中的分子进行优化,以进行属性预测。为了创建一个更平滑的环境来执行优化,使用高斯过程模型来建模属性预测器模型。训练高斯过程的2000个分子被选择为最大限度地多样化。在潜在空间中进行优化,以找到一个最大化目标分子
选择优化的目标是5 × QED - SA,其中QED为药物相似性的定量估计,38,SAS为合成可及性评分这一目标代表了一个粗略的估计,即找到最像药物的分子,也很容易合成。
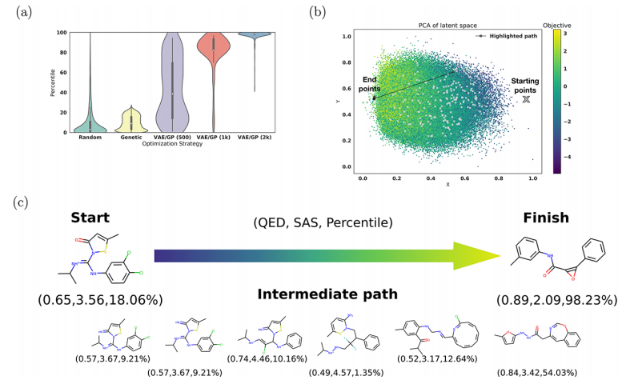
图4a显示了使用高斯过程(GP)模型对潜在空间表示的优化始终导致分子具有更高的百分位数。
图4b显示了二维PCA表示中从起始分子到最终分子的一次优化路径,最终分子位于高目标值区域。
图4c显示了使用高斯插值沿此优化路径解码的分子。
在使用1000个分子训练的GP模型上执行此优化,可以得到稍宽的分子范围,如图4a所示。由于训练集较小,GP的预测能力较低,在潜在空间中进行优化时,GP会优化到几个局部最小值,而不是全局优化。在难以完全描述分子中所需的所有特征的情况下,使用这种局部优化方法来获得更大的潜在分子多样性可能会更好。
由于文章比较老,当时2017年投稿的,VAE还是火的时候,想法很好,加一个MLP约束分子的潜在分布。