最近,我发现学徒在学习GEO数据挖掘的过程中,遇到了第一个也是至关重要的一个难题就是对下载后的数据集进行合适的分组,因为只有对样本进行合适的分组,才有可能得到我们想要的信息。但是不同的GSE数据集有不同的临床信息,那么我们应该挑选合适的临床信息来进行分组呢? 这里面涉及到两个问题,首先是能否看懂数据集配套的文章,从而达到正确的生物学意义的分组,其次能否通过R代码实现这个分组。同样的我也是安排学徒完成了部分任务并且总结出来了! 下面看学徒的表演(PS: 图片较多的推文,排版真的是吓死人!)
Jimmy大神怎么说过,只有多做、多错,才能真正的掌握。所以下面通过几个实战来说明。
- 首先是通过对一篇文献Identification of potential core genes in triple negative breast cancer using bioinformatics analysis所用到的三个TNBC(Triple-Negative Breast Cancer)三阴性乳腺癌的三个数据集:GSE38959、GSE45827以及GSE62194进行分组,首先对GSE38959进行分析。
library(GEOquery)
# 这个包需要注意两个配置,一般来说自动化的配置是足够的。
#Setting options('download.file.method.GEOquery'='auto')
#Setting options('GEOquery.inmemory.gpl'=FALSE)
gset <- getGEO('GSE38959', destdir=".",
AnnotGPL = F, ## 注释文件
getGPL = F) ## 平台文件
class(gset) #查看数据类型
length(gset) #
class(gset[[1]])
# 因为这个GEO数据集只有一个GPL平台,所以下载到的是一个含有一个元素的list
a=gset[[1]] #
dat=exprs(a) #a现在是一个对象,取a这个对象通过看说明书知道要用exprs这个函数
dim(dat)#看一下dat这个矩阵的维度
dat[1:4,1:4] #查看dat这个矩阵的1至4行和1至4列,逗号前为行,逗号后为列
pd=pData(a) #通过查看说明书知道取对象a里的临床信息用pData复制
pd就是这个数据集的临床信息,查看后如下
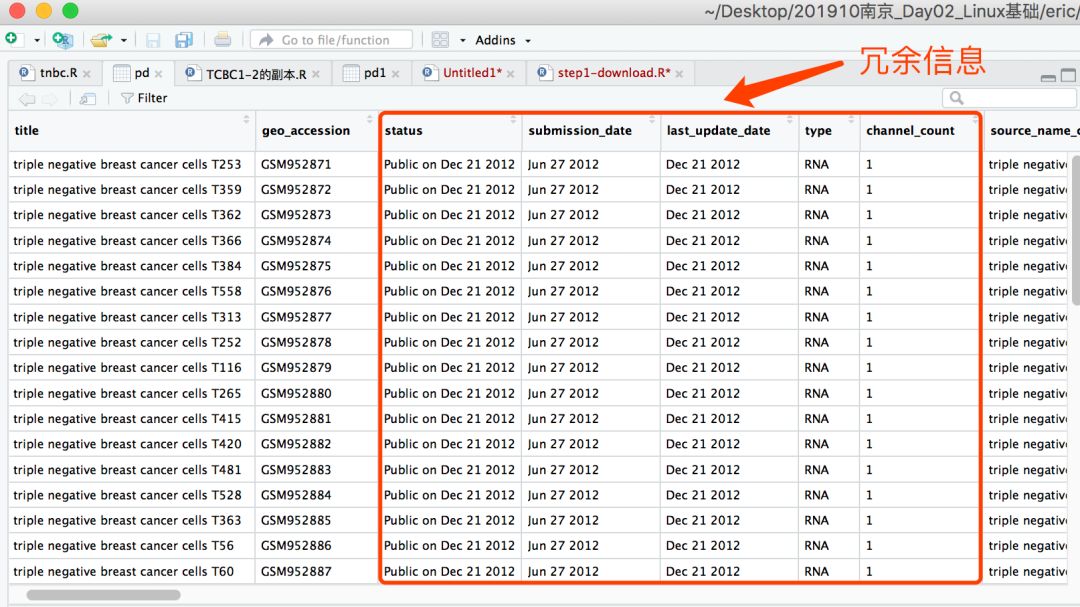
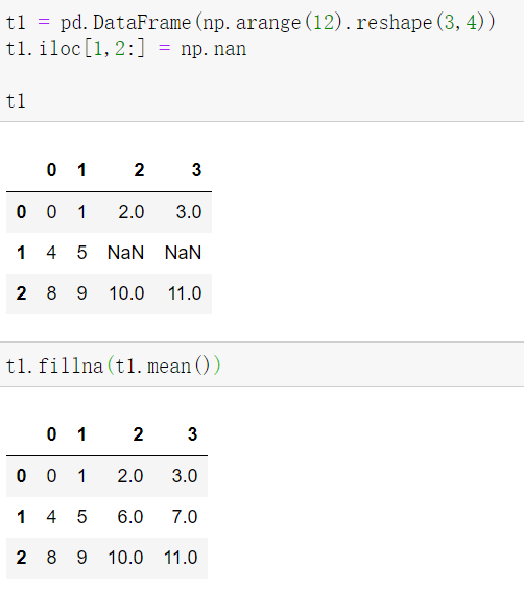
会发现有些信息是冗余的,有些是有效信息可以用来分组,但是表型记录太多,看起来会混淆,所以需要去除那些冗余信息,就是在所有样本里面表型记录都一致的列。如何去冗余,见原文对表型数据框进行去冗余。
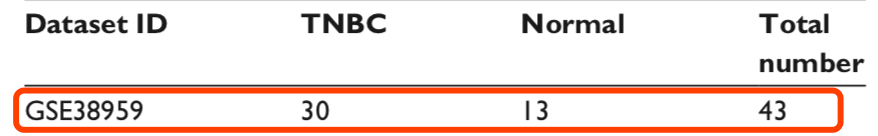
阅读文章后发现原文把样本分为2组:肿瘤与正常,而且总共只有43个样本,而临床信息有47个样本,说明有效信息列含有3个或3个以上元素,可以再缩小范围。(注意!如果样本数刚好去冗余就行!)
pd1=pd[,apply(pd, 2, function(x){
length(unique(x))>2})] #缩小范围
dim(pd1)
#[1] 47 9复制
发现范围已经缩小很多。对数据框再用apply循环去查找文章作者是用哪一列来分组的
apply(pd1,2,table)复制

通过循环,就可以清楚的知道该用哪一列来进行分组啦
然后是搜索关键字进行分组
TNBC=rownames(pd1[grepl('triple negative breast cancer cells',as.character(pd$`cell type:ch1`)),]) #肿瘤组
NOR=rownames(pd1[grepl('mammary gland ductal cells',as.character(pd$`cell type:ch1`)),]) #正常组
dat=dat[,c(TNBC,NOR)] #对表达矩阵取子集
group_list=c(rep('TNBC',length(TNBC)) ,
rep('NOR',length(NOR))) #分组信息
table(group_list)
#group_list
#NOR TNBC
#13 30 复制
第二个数据集GSE45827同样的方法,重复的地方不赘述,从有差异的地方开始。

文中为了2组,用了52个样本。

然后是搜索关键字进行分组
TNBC=rownames(pd[grepl('Human Basal Tumor Sample',as.character(pd$source_name_ch1)),]) #肿瘤组
NOR=rownames(pd[grepl('Human Normal',as.character(pd$source_name_ch1)),]) #正常组
dat=dat[,c(TNBC,NOR)]
group_list=c(rep('TNBC',length(TNBC)),
rep('NOR',length(NOR))) #分组信息
table(group_list)
#group_list
#NOR TNBC
# 11 41复制
第三个数据集同理
2.选取OTSCC(Oral Tongue Squamous Cell Carcinoma)口腔舌鳞癌Integrative analysis of gene expression profiles reveals distinct molecular characteristics in oral tongue squamous cell carcinoma的GSE13601, GSE31056 and GSE78060三个数据集
这里主要说一下GSE31056这一个数据集,需要一定的背景知识与细心才能正常分组,原文里

如果用我们之前的方法找是找不到的,因为细心点你会发现GSE给的位置不止tongue,还有mouth等,而文章只需要tongue。所以我们需要对数据集取子集。
pd1=pd[,apply(pd, 2, function(x){
length(unique(x))>1})]
pd1=pd1[grepl('tongue',as.character(pd$characteristics_ch1.2)),]
apply(pd1,2,table)复制

根据生物学相关知识,margin其实就属于normal,所以就找出了分组。
所以可以看到生物学知识多么重要:没有生物学背景的数据分析很危险
TU=rownames(pd1[grepl('tumor',as.character(pd1$`site:ch1`)),]) #肿瘤
NOR=rownames(pd1[grepl('margin',as.character(pd1$`site:ch1`)),])#正常
dat=dat[,c(TU,NOR)]#取子集
group_list=c(rep('TU',length(TU)), #分组信息
rep('NOR',length(NOR)))
table(group_list)
#group_list
#NOR TU
# 39 12 复制
3.分析关于ccRCC (clear cell renal cell carcinoma)肾透明细胞癌的一个GSE子集GSE53757
下载数据、提取表达矩阵与临床信息方法与前面一直,这里就不赘述,也是从有差异的地方开始。这里文章作者只需要第三期的正常与肿瘤组的样本
table(pd$`tissue:ch1`)
table(pd$`tumor stage:ch1`)
table(pd$source_name_ch1)复制

通过table函数,我们看到总共144个样本,其中有72个正常与72个肿瘤样本;第三期肿瘤和正常样本总各有14个,下面我们就需要提取我们需要的数据
patient_t = pd[pd$`tissue:ch1`=='clear cell renal cell carcinoma' &
pd$`tumor stage:ch1`=='stage 3', ] #通过向量取交集的方式来取
patient_n=pd[pd$source_name_ch1=='normal match to Stage 3 ccRCC',]
colnames(dat)=as.character(pd[,1]) #表达矩阵的列名就是临床信息的第一列
dat=dat[,c(patient_t[,1],patient_n[,1])]#取子集
group_list=rep(c('ccRCC','normal'),each=14) #分组信息
table(group_list)
#group_list
#ccRCC normal
#14 14 复制
总结一下,我们可以根据自己的需求选取合适的代码去进行有效的分组,在不同的情况下选取最合适当下的方法,方便自己去做后续的数据分析。