谷禾健康

目前,原核生物的系统发育分类依赖于16S rRNA基因序列,这种序列在细菌中广泛存在且高度保守,但即使16S rDNA测序的结果显示某些细菌间序列相似性≥99%,但它们仍然不能被归为同一物种。因为基因序列中的一些保守区域并不一定代表真正的进化历史,水平基因转移也可能会影响分析结果。
为此研究人员开发了一种方法,并为该方法创建了一个web应用程序Phy5,以及它的命令行版本Phy5cli。
该方法旨在对任何物种或亚种进行系统发育评估,包括与已知物种的基因序列相似性非常低的非致病菌株,可以轻松地进行系统发育树分析。
文中以E.coli、Shigella、Yersinia、Klebsiella和Neisseria spp.的系统发育关系为例进行了应用与验证。
方 法
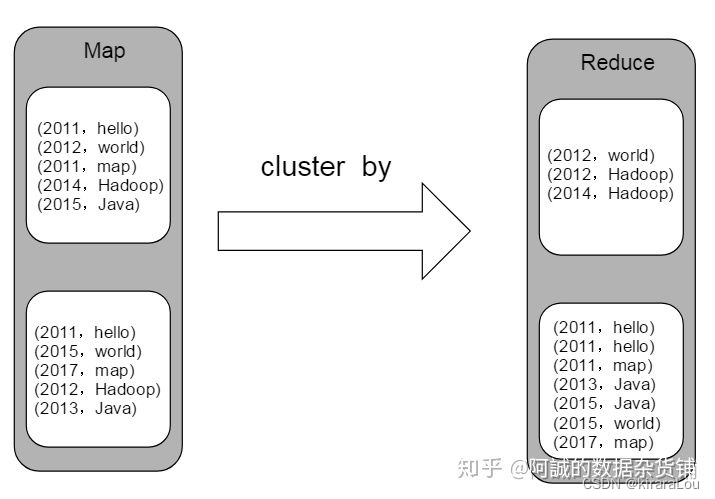
通过分析短核苷酸的频率差异来区分高度相似物种,划分成不同的类群,从而推断它们在系统发育上的关系。
研究使用R v4.12和Biostrings软件包分别对每个细菌样品的三核苷酸、四核苷酸、五核苷酸和六核苷酸频率进行了测定。
这些K-mer(由k个连续核苷酸组成的单元)频率是构建系统发育树的重要信息。通过测定K-mer频率,可以使用层次聚类分析(hierarchical cluster analysis)将样品细菌基因组的相似性进行比较。
在此过程中,使用曼哈顿距离(Manhattan)估计样本间的差异程度,并使用沃德算法(Ward’s)来判断样本间的相似性和关联性。
在该方法中,将互补的K-mer对(例如AAA vs TTT)视为相同的核苷酸序列,以降低这种互补配对对结果的影响。如下图,就是基于五核苷酸频率的系统发育树构建方案。

应用与验证结果
1
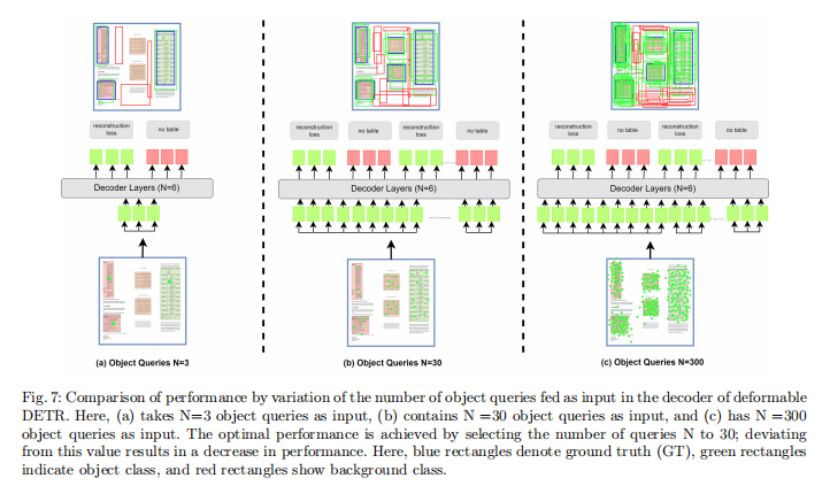
分别基于五核苷酸频率分析和MLST构建的110个Yersinia菌株的系统发育树
如下图所示,图A为基于五核苷酸频率分析所构建的系统发育树,采用曼哈顿距离和Ward算法。
图B是采用neighbor-joining方法的多重位点序列分型(MLST) 分析所构建的系统发育树。

节点上的数字表示在1000个bootstrap值中出现的百分比。颜色区分不同种水平分类。
结果表示两种方法在这个特定的数据集上所产生的系统发育树是高度相似的,它们都可以很好地区分Yersinia和其他物种。
而基于16S rRNA基因序列分析所构建的系统发育树(MAFFT工具)无法区分这些物种,如下图所示,下图B为基于16S rRNA基因序列分析所构建的系统发育树。

2
基于五核苷酸频率分析方法构建系统发育树可以应用于任何物种,且仅需要通过高通量测序技术获取的短链
如下图,根据五核苷酸频率构建了E. albertii、E. coli和Shigella的系统发育树。

发现E. albertii 和致肠出血性大肠杆菌菌株(包括O157、O121和O111等)密切相关,并与Shigella和非致病性E. coli(如K-12菌株)分开排列。
Shigella菌株与肠侵袭性大肠杆菌(EIEC)可以区分开来,并与其他大肠杆菌菌株形成一个分支。
3
基于五核苷酸频率分析方法构建的系统发育树仅适用于近亲物种
研究人员从GenBank序列数据库下载了24个Ipomoea属植物的30个叶绿体基因组的核苷酸序列,并使用五核苷酸频率分析方法构建系统发育树,结果如下图,只构建出来与Ipomoea相关的物种。

如其它隶属于Ipomoea的物种并没有被发现。
研究人员还测试了使用五核苷酸频率分析方法构建各种远亲物种的系统发育树,包括嗜热古菌和细菌。

但结论与上面一致。这个方法无法准确地反映远亲物种之间的亲缘关系。
结 论
基于五核苷酸频率分析的系统发育分析是一个具有潜力的微生物系统发育分析的方法。该方法对于近亲物种表现良好,对于远亲物种,能力还不够。
研究人员将该方法制作成了Phy5的R包,工具小巧,运行速度也快,可在这里获得:
也可以访问在线版本:
参考文献:
Nakano Y, Domon Y, Yamagishi K. Phylogenetic trees of closely related bacterial species and subspecies based on frequencies of short nucleotide sequences. PLoS One. 2023 Apr 20;18(4):e0268847. doi: 10.1371/journal.pone.0268847. PMID: 37079522; PMCID: PMC10118083.