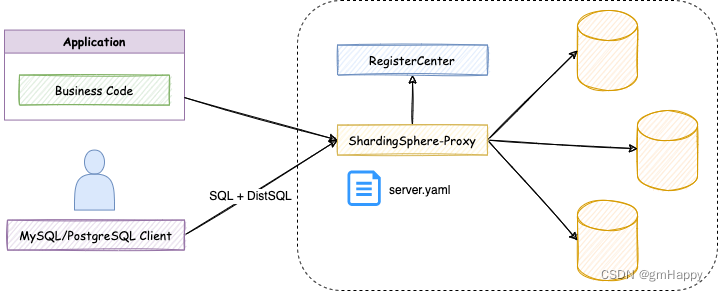
AMBER分子动力学模拟之结果分析(最低能量结果)-- HIV蛋白酶-抑制剂复合物(3)
在analysis目录下
解析.out文件
下载process_mdout.perl 脚本
perl process_mdout.perl ../md/md0.out ../md/md1.out ../md/md2.out # 可以不使用md0.out
# 或者
$AMBERHOME/bin/process_mdout.per ../md/md0.out ../md/md1.out ../md/md2.out
解析出如下文件
可视化
如果没有显示器,在命令窗口显示作画,可以使用以一下软件:Xming和Xshell,
Xshell的通道需要开启:

xmgrace summary.TEMP
模拟跑的不好!!!仅供参考
最低能量
可以定位具有最低能量的结构,我们可以通过查看process mdout.perl生成的summary.EPTOT文件来定位最低能量。但首先我们需要剥离数据的前50ps,因为这代表加热阶段。这是删除了此部分的文件。(下面的awk 脚本会为我们快速找到文件中的最小值。每次找到新的最小值时,它都会打印一个值,最后打印的值将是找到的最低能量。
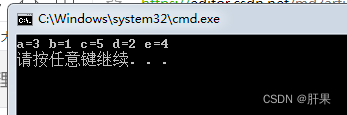
cat summary.EPTOT | awk '{if($2 <min) {min=$2; print $1" "min}}'

注意:如果您自己进行模拟,您的答案可能会有所不同因为机器精度的变化意味着您将探索略有不同的相空间区域。
找能量最低的结构
因此,最低能量为-547.4913kcal/mol,它发生在我们的模拟中42.505ns。我们可以使用grep找出它在输出文件中的位置:
grep 42505.000*.out从所有的out文件中检索42505.000
注意:42505.000是process_mdout.perl解析的所有时间之和,帧位置在哪个位置需要重新计算
所以它发生在我们第9个平衡步骤的第1227500步。我们在平衡期间每500步(ntwx=500)写入nc文件,因此这应该代表xx.crd的第2455帧(1227500/500)。让我们使用cpptraj提取该结构并查看它。
vim analysis.sh
#!/bin/sh
parm ../top/com.top
trajin ../md/md2.crd 107 107 1 # 需要自己计算帧的位置
reference ../top/com.pdb
center :1-199 mass # 参考残基
image center familiar
rms reference @CA
trajout md_low.pdb pdb
$AMBERHOME/bin/cpptraj -i anaylsis.sh