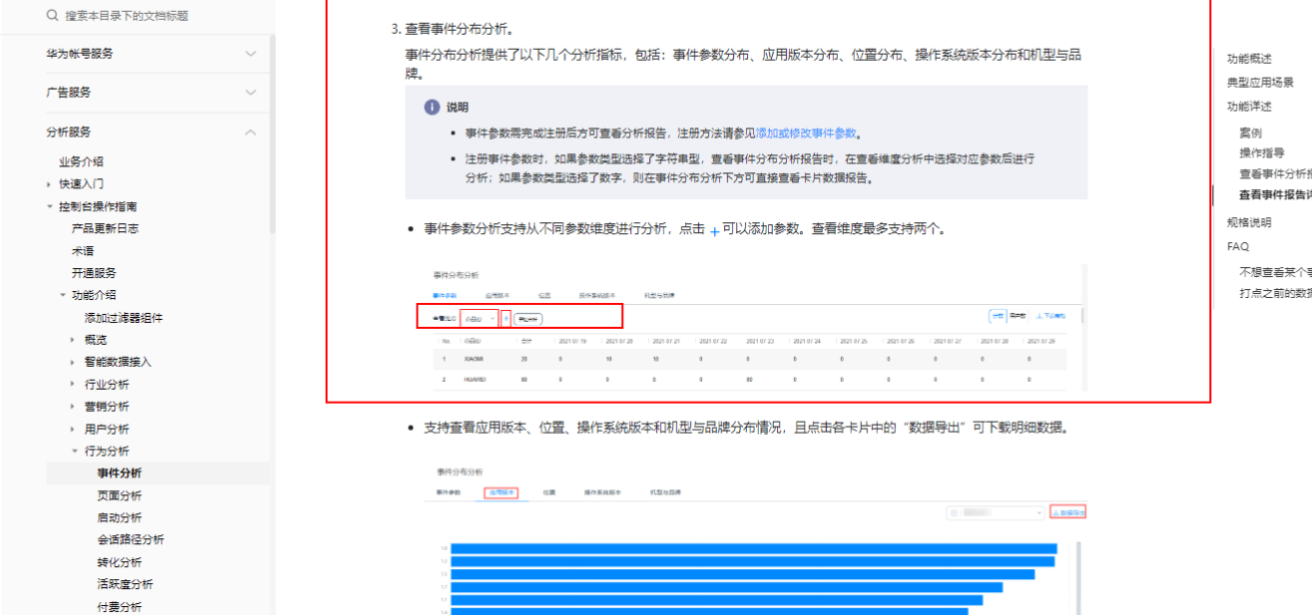
用交联质谱和共分离质谱识别PPI,并用AlphaFold-Multimer预测蛋白相互作用复合体的结构
期刊介绍
这篇文章是2023年2月23日发表在《molecular systems biology》上的一篇结构生物学文章,这个期刊的影响因子为7.1。

作者介绍
左下图为1985年的弗雷德里克癌症研究中心。1969年,在尼克松总统签署了《禁止生物武器公约》后,位于马里兰州弗雷德里克的德特里克堡就从美国生物战争研究中心变成了癌症研究中心。图片由美国国家肿瘤研究所提供。

背景和技术
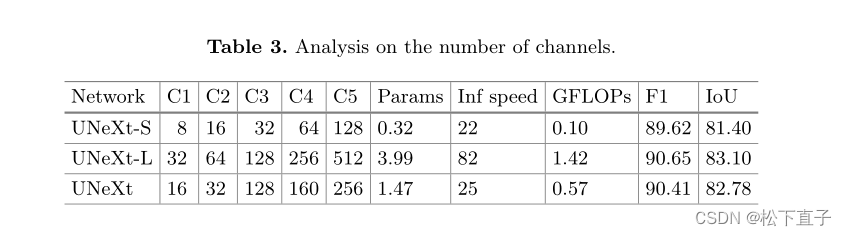
PPI概述
生命依赖于生物大分子之间的功能性相互作用,其中蛋白质之间的相互作用(Protein-Protein Interaction,PPI)是数量最多、最多样化的。我们之前也学过了PPI,它们的结构和功能密切相关,但是阐明这些结构很困难。蛋白质组学和遗传学方法已经得到了大量的PPI数据,但是对其拓扑结构的了解很少。如果我们能够获得这些蛋白质以及它们相互作用的复合体的结构,那么就能揭示以前没有过的生物知识,为生命科学做出重大贡献。
那么如何得到蛋白质相互作用复合体的结构呢?
AlphaFold-Multimer预测复合体结构
我们知道AlphaFold可以预测蛋白质结构,那么复合物也可以被预测吗?当然可以,不过是用的AlphaFold Multimer。AlphaFold Multimer是一种基于AlphaFold 2.3.2的扩展,专门用于预测蛋白质复合物的结构。它重新训练了神经网络,考虑了复合物的对称性、配对的多序列比对(MSA)、残基选择和位置编码。它可以更准确地预测复合物的结合界面,并在同源和异源复合物上都有很好的表现。它的代码和权重已经开源,也可以通过Colab notebook在线使用。
AlphaFold Multimer使得精准预测蛋白质复合物成为可能。这使得在相互作用组(interactomes)中建立结构-功能之间的关系成为可能,可以更加深入地了解蛋白质,开启了结构系统生物学的新时代。

研究对象:枯草芽孢杆菌(B.subtilis)
本文的研究对象是枯草芽孢杆菌,目的是探索它的蛋白质相互作用网络(PPI)。枯草芽孢杆菌是一种革兰氏阳性、好氧、能形成抗逆性孢子的细菌,广泛分布于土壤和植物表面。它不仅是商业生产蛋白质的重要工具菌,也是人类重要病原菌炭疽杆菌、单核增生李斯特菌和金黄色葡萄球菌的近缘物种。作为芽孢杆菌科(Bacilaceae)的模式菌株,它在生物学和医学上具有重要的研究价值。然而,迄今为止,对枯草芽孢杆菌的PPI进行系统性的筛选和分析还很少见。
预测哪些PPI的结构
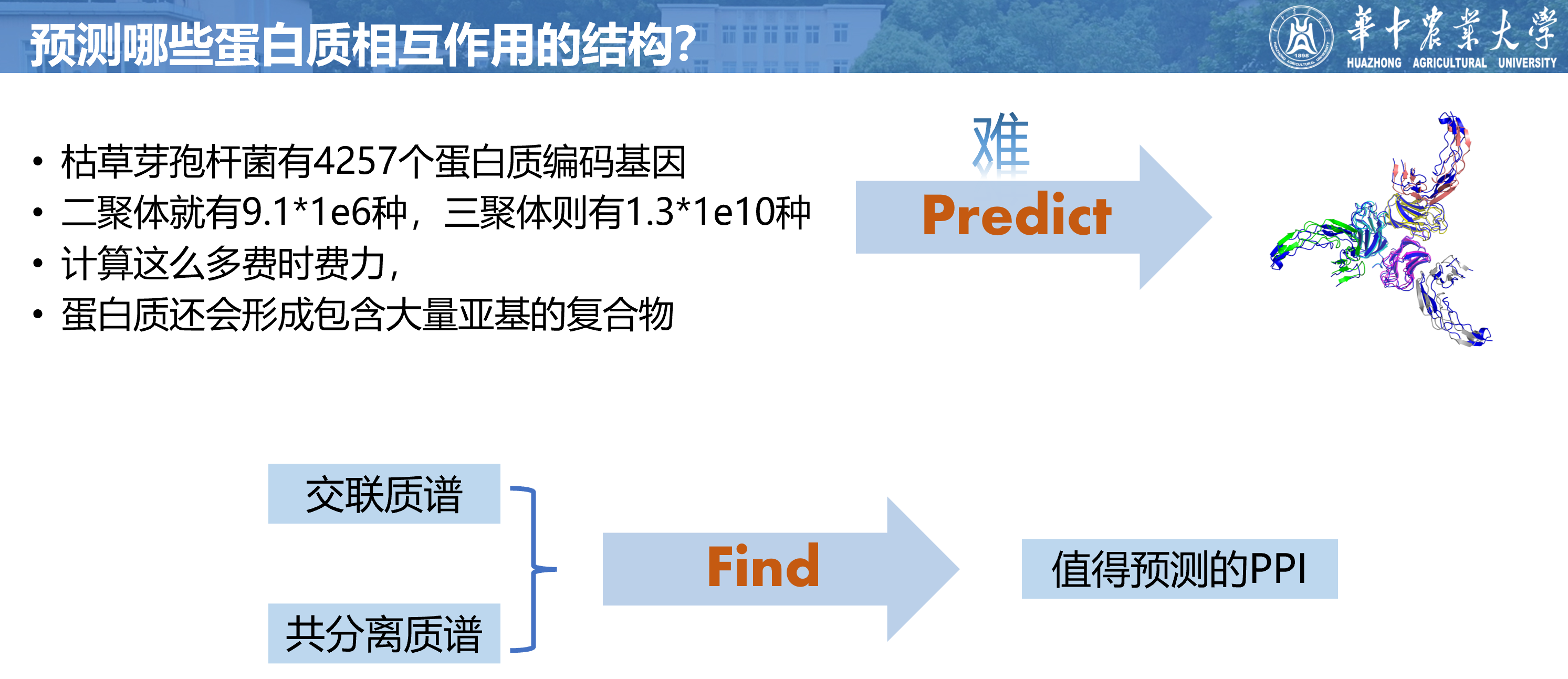
我们知道了如何预测相互作用的结构,那预测哪些相互作用呢?枯草芽孢杆菌有4257个蛋白质编码基因,二聚体就有 C 4257 2 = 9.1 × 1 0 6 C_{4257}^2=9.1\times10^6 C42572=9.1×106种,三聚体则有 C 4257 3 = 1.3 × 1 0 10 C_{4257}^3=1.3\times10^{10} C42573=1.3×1010种,计算这么多费时费力,而且蛋白质还会形成包含大量亚基的复合物,因此搞清楚预测哪些PPIs也是一个问题。
作者为了解决这个问题,就是用交联质谱和共分离质谱找到哪些PPI值得预测。
接下来介绍介绍什么是质谱、交联质谱和共分离质谱。
主要技术介绍
这篇文章的核心就是这两个质谱技术,我们看看这个质谱技术是什么?
质谱(Mass Spectrometry)
质谱其实在高中学过,类似于核磁共振氢谱,就是用电场和磁场将运动的离子按照,通过质荷比分离后进行检测,可以测量分子的质量和结构。在蛋白质组学中,质谱可以用来鉴定蛋白的序列、纯度、分子量、翻译后修饰等信息。
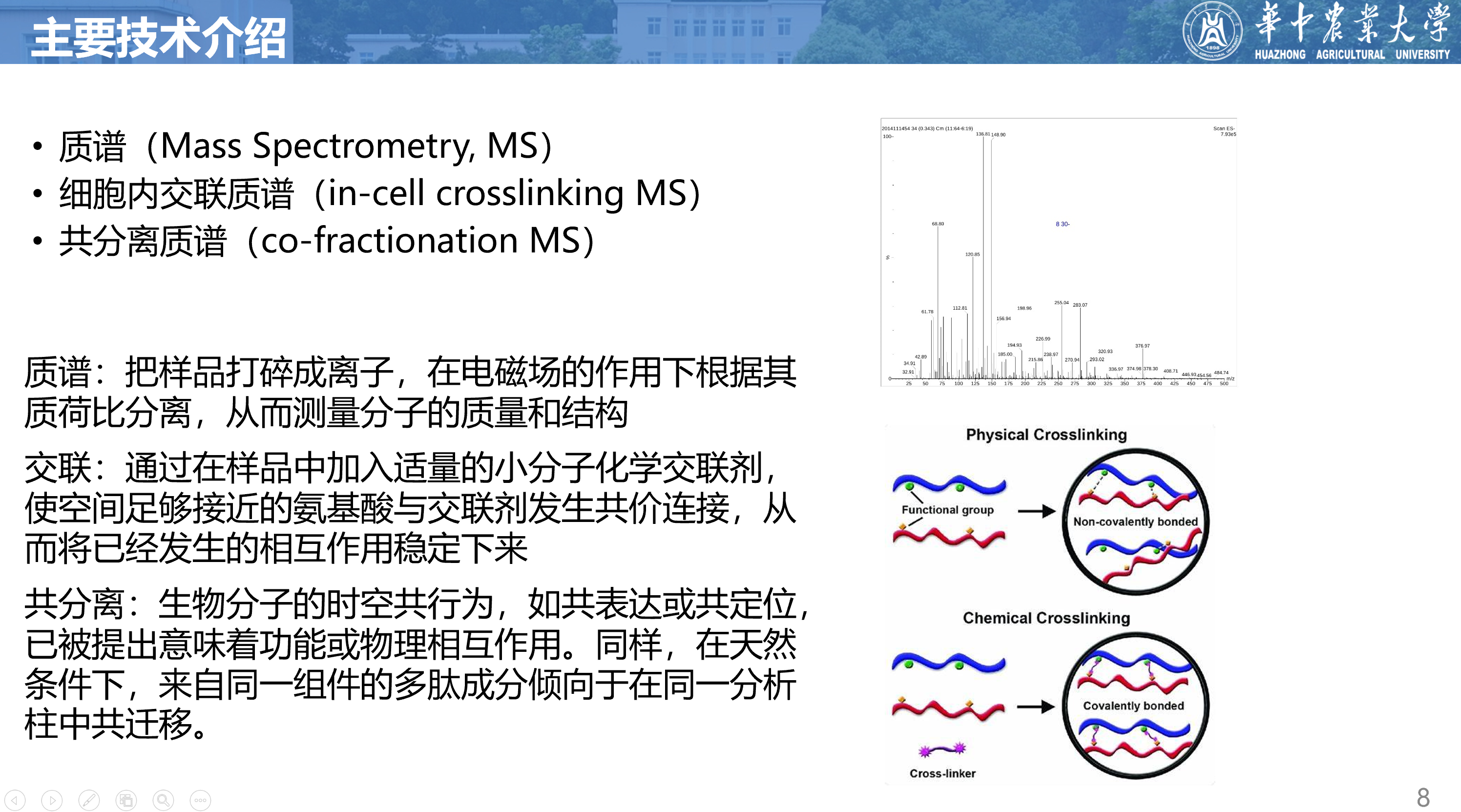
交联(Crosslinking)
交联反应在近端残基之间增加了新的共价键。交联质谱技术(Cross-linking mass spectrometry,XL-MS)是将蛋白质化学交联技术与质谱技术相结合,用于研究蛋白质结构以及蛋白质相互作用的新方法。通过在样品中加入适量的小分子化学交联剂,使空间足够接近的氨基酸与交联剂发生共价连接,从而将已经发生的相互作用稳定下来,通过质谱与交联质谱数据处理软件解析确定发生相互作用的蛋白质以及具体作用的交联位点,获取蛋白质相 互作用信息,结合交联剂的长度对蛋白质的空间结构 及蛋白质复合体亚基的空间排列进行预判。
共分离(Co-fractionation)
如果我们在校园里看到两个人吃饭在一起、学习也在一起,那么他们俩之间很可能有相互作用,有可能是情侣或是好基友。生物分子也是一样的。
生物分子的时空共行为,如共表达或共定位,已被提出意味着功能或物理相互作用。同样,在天然条件下,来自同一组件的多肽成分倾向于在同一分析柱中共迁移。因此,具有相似共分离谱的蛋白质可能提示明显的共定位。这种相关关系最初用于细胞器蛋白质组学,使用密度梯度离心进行生化分级,但这一概念被外推到相互作用蛋白质组学,从而产生了共分离-MS(coFrac-MS)。
由于完整复合物的亚基倾向于共分离,因此可以使用分离谱之间的相关性作为核心重要性的特征,从这些数据中预测蛋白质复合物。
研究目标
在前文中,我们介绍了ppi的概念,以及如何利用AlphaFold Multimer预测蛋白质复合物的结构。我们还介绍了我们的研究对象——枯草芽孢杆菌,以及两种用于识别PPI的质谱技术:交联质谱和共分离质谱。本文的研究目标是:结合这两种质谱技术,识别枯草芽孢杆菌中的PPI,并用AlphaFold Multimer预测相应的蛋白质复合物结构。
具体流程
交联质谱找PPI
本文采用了两种质谱技术来识别ppi,第一种是交联质谱。
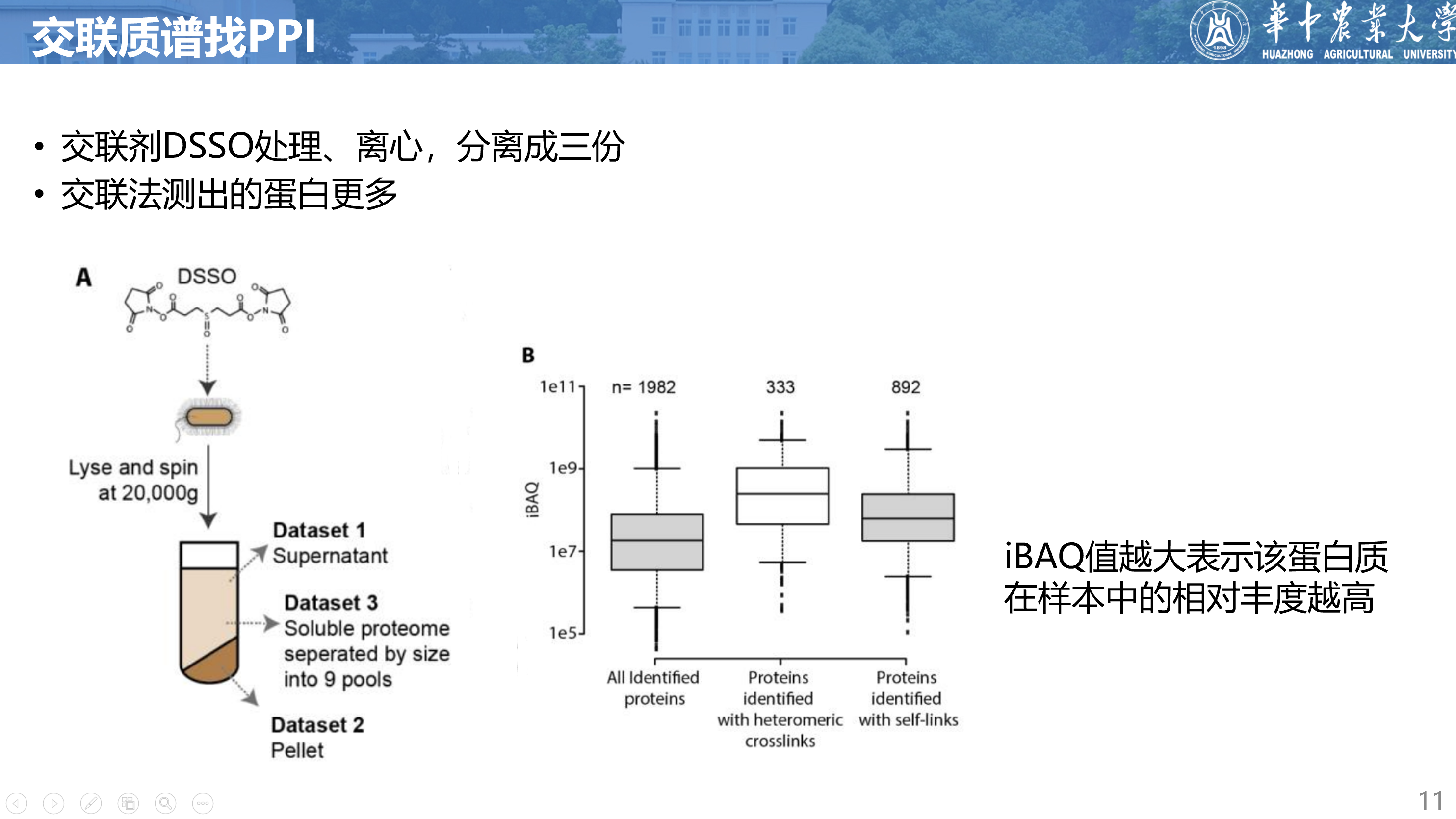
作者首先用可穿透细胞膜的交联剂DSSO将B. subtilis细胞中的蛋白质交联起来。然后,细胞被破碎并提取蛋白质,再用胰蛋白酶切割成肽段。这些肽段经过阳离子交换和尺寸排除色谱分离后,进行质谱分析和数据库匹配,得到三个数据集。
对每个数据集进行FDR校正后(FDR率为2%),共得到560个ppi,由1268个残基对支持。这些ppi涉及337个不同的蛋白质,另外还有629个蛋白质只出现在自连接中。这些蛋白质占了全细胞分析中检测到的1982个蛋白质的一半。
iBAQ(intensity-based absolute quantification)是一种根据每个蛋白质的质谱图强度计算的定量指标,反映了蛋白质在样本中的相对丰度。iBAQ值越高,蛋白质的相对丰度越高。因此,可以用iBAQ值来比较不同样本或不同蛋白质之间的表达水平。
从右图可以看出,交联法检测到的蛋白质的iBAQ值比所有检测到的蛋白质的iBAQ值高一个数量级,所以交联法测出的蛋白更多。
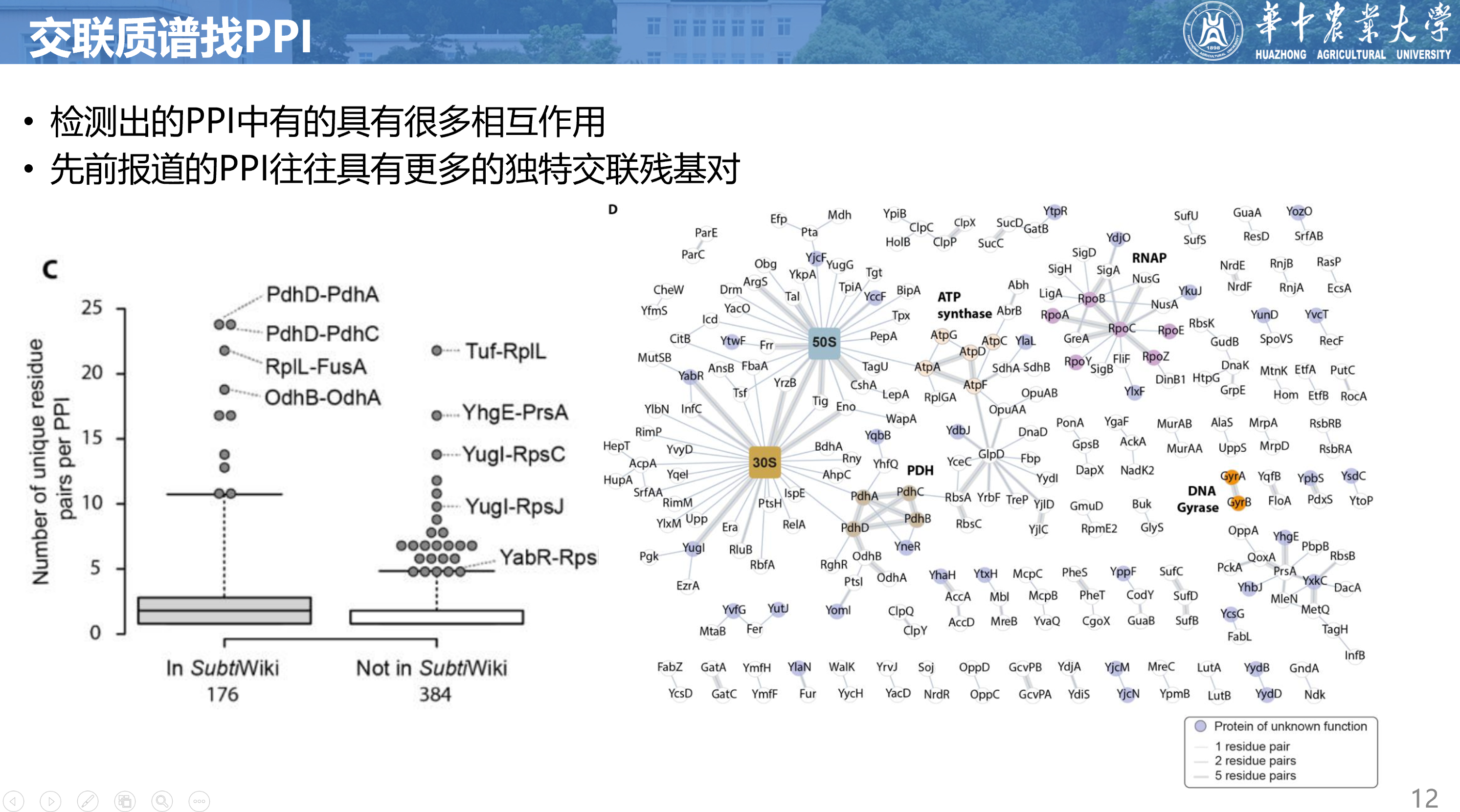
在交联法检测到的560个ppi中,有176个已经在SubtiWiki中有报道。这个ppi网络中有一些高丰度的蛋白质,它们与多个蛋白质发生交联,例如核糖体蛋白L7/L12 (RplL)、L1 (RplA)和RS3 (RpsC),延伸因子Ef-Tu (TufA)和Ef-G (FusA),以及RNA伴侣蛋白CspC和CspB,它们分别与20多个蛋白质形成交联。除了CspB,这些蛋白质的强度都排在前30位,其中Ef-Tu (TufA)和RplL是最高的(Dataset EV2)。如果排除这些蛋白质的交联,那么剩下的310个ppi中有186个是新发现的。
与先前报道的ppi相比,新发现的ppi往往具有较少的独特交联残基对,这可能意味着细胞中这些相互作用/蛋白质复合物的丰度较低。
右图展示了得到的PPI网络,其中蓝色的节点表示未被表征的蛋白质,也就是结构未知的蛋白质。
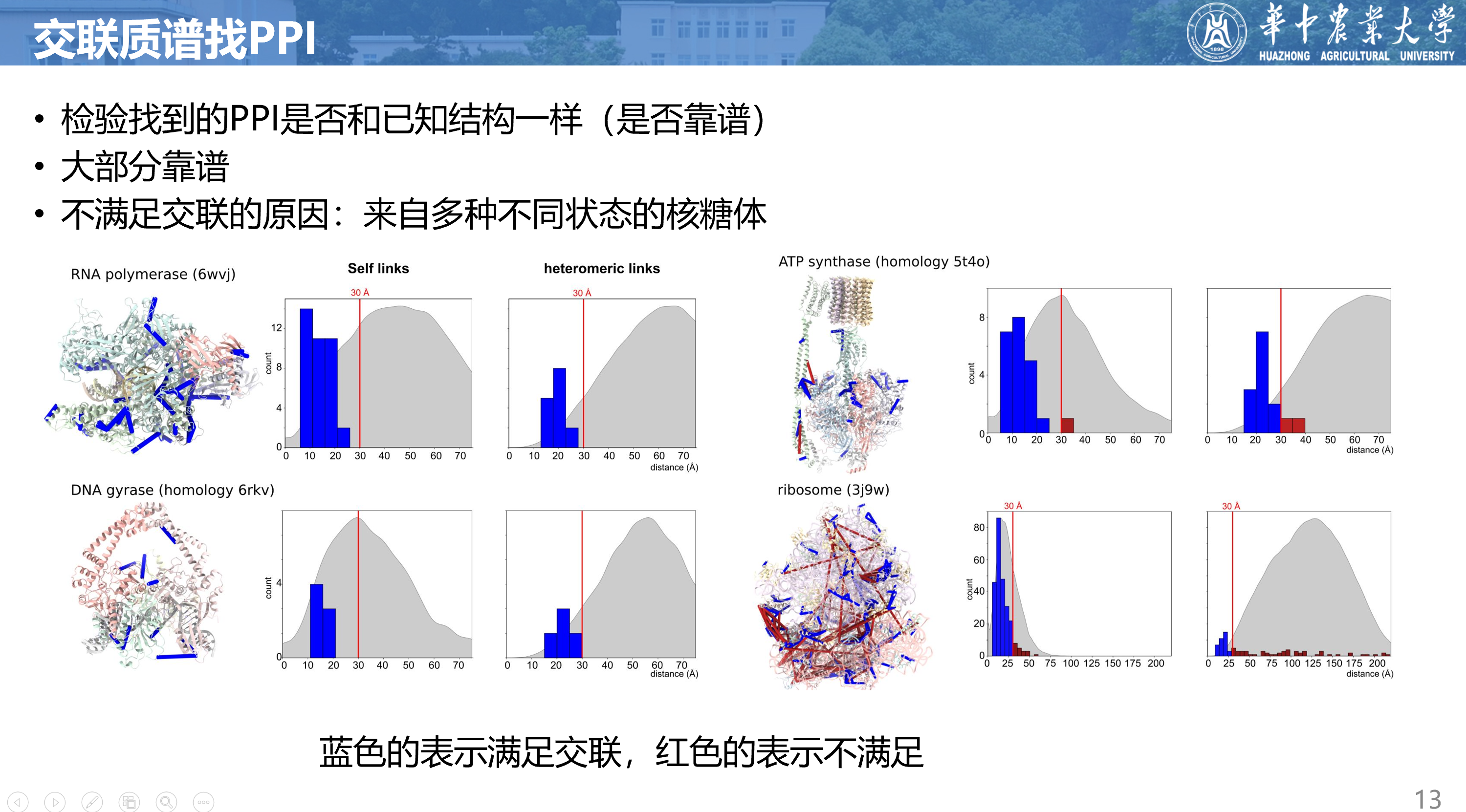
为了验证ppi的可靠性,我们把数据集中的交联数据映射到枯草芽孢杆菌RNA聚合酶的已知结构,以及DNA回转酶(gyrase)和ATP合酶的同源模型上。
从下图可以看出,蓝色的线表示交联数据与结构模型一致,红色的线表示交联数据与结构模型不一致。大部分的交联数据都是与结构模型相符的,这说明检测结果具有较高的可信度。
然而,在最下面的核糖体中,有很多红色的线(74/343,21.5%),表示交联数据与结构模型不符。作者认为这些交联可能来自于细胞中存在的多种不同状态的核糖体,包括多核糖体相互作用和核糖体前组装。
此外,作者还发现在许多与核糖体亚基交联的蛋白中,有两个蛋白与多个30s蛋白形成了密切的交联。这两个蛋白是:Yugl与8个30s蛋白共有34个交联点,YabR与4个30s蛋白共有10个交联点。
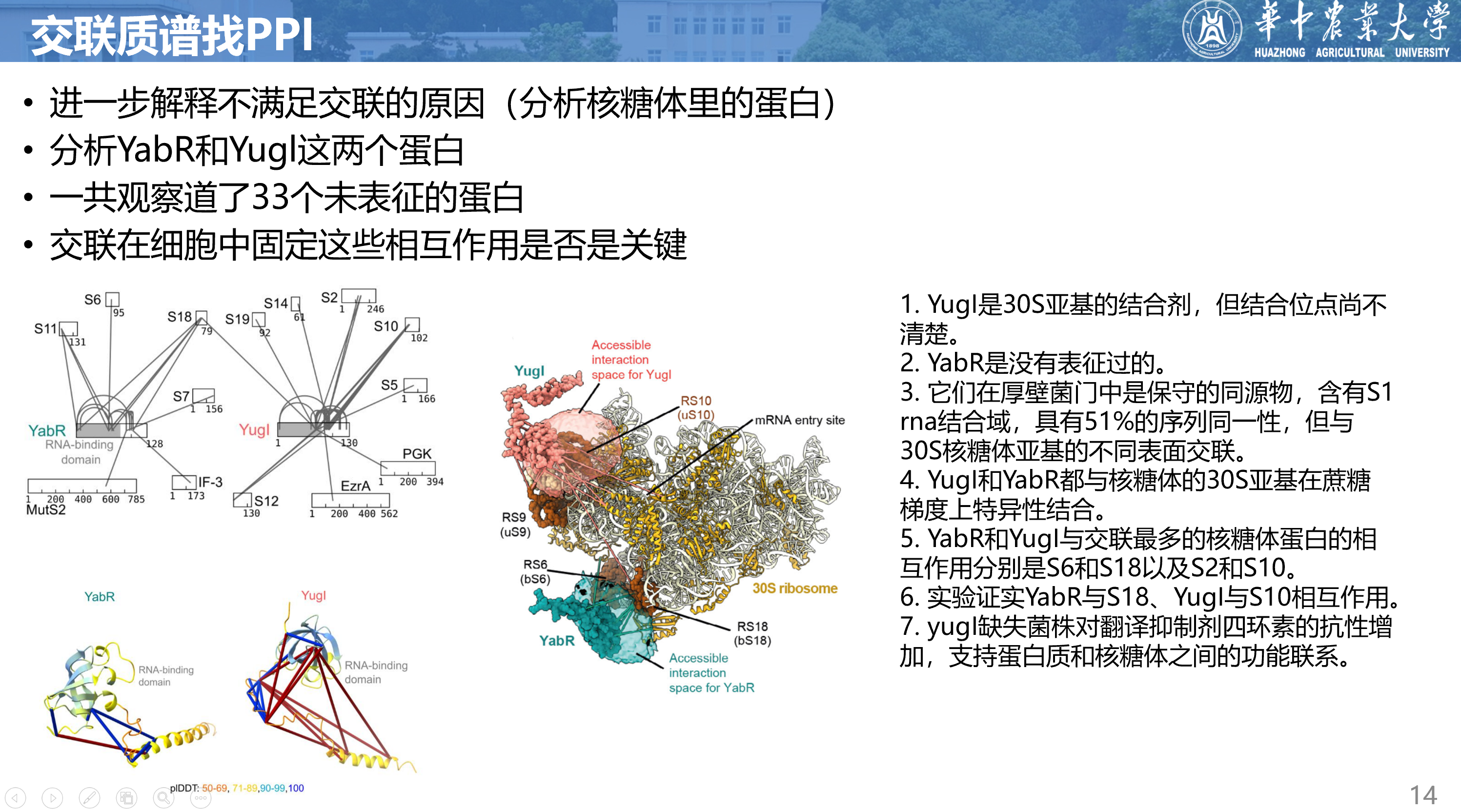
除了YabR,我们的交联质谱网络中还有33个未表征的蛋白质。我们想知道为什么我们能在以前遗漏的ppi中发现它们。为了探究交联是否是固定这些相互作用的关键因素,我们接下来比较了未处理和交联的细胞裂解液中哪些蛋白质复合物能通过大小排斥色谱(SEC)分离。
共分离质谱找PPI
接下来,我们进入第二个质谱的环节——共分离质谱。回顾一下,生物分子的时空共行为,如共表达或共定位,被认为是功能或物理相互作用的线索。同理,在天然条件下,来自同一组件的多肽成分会在同一分析柱中共迁移。因此,共分离谱相似的蛋白质可能暗示着明显的共定位。
下面这个图就是为了说明交联能稳定相互作用,使得复合物一起被洗脱。图中A有两行,上面是未处理过的,下面是交联处理的,可以看到交联处理后的曲线波形更加一致,波峰和波谷都能对应上。图中B则可以根据曲线的阴影来判断,阴影表示标准差,橙色的交联处理后的标准差更小,也说明了共洗脱的现象。
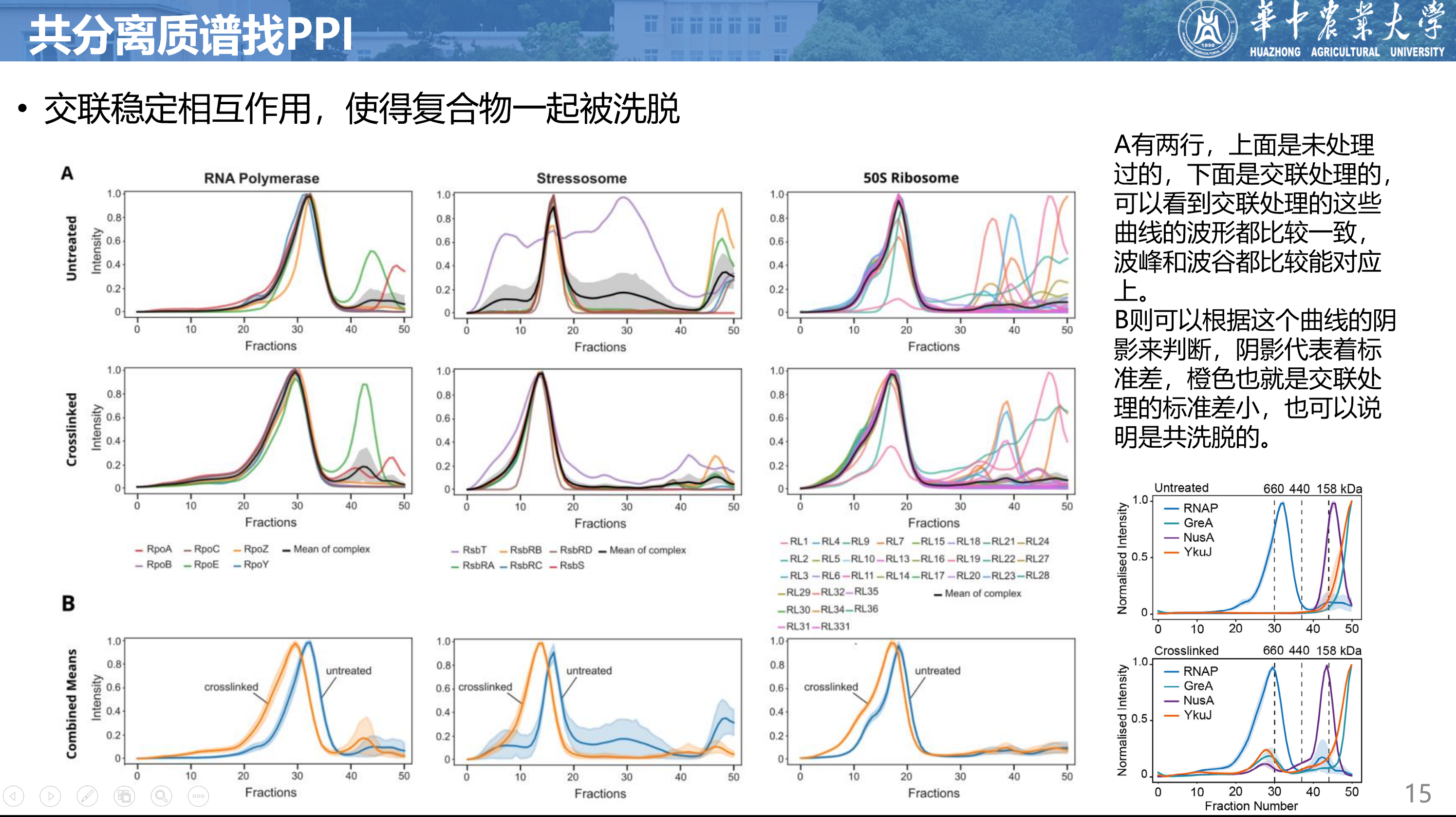
作者分析了交联质谱的PPI在共分离质谱中的表现。在排除核糖体对后,265个PPI中有109个在交联前后都显示出相似的共洗脱行为。在剩下的156个PPI中,超过2/3的在交联后的共洗脱评分更高。
从下图可以看出,大部分的点位于左上角,这说明大部分的PPI在交联后的共洗脱分数更高。反之,共洗脱的蛋白质也可以被视为潜在的PPI。
总之,在细胞裂解之前稳定蛋白质复合物,对于识别未表征蛋白质的相互作用是非常有效的。交联使得未表征蛋白质在cofrc-ms数据中的已鉴定PPI从36%增加到64%。
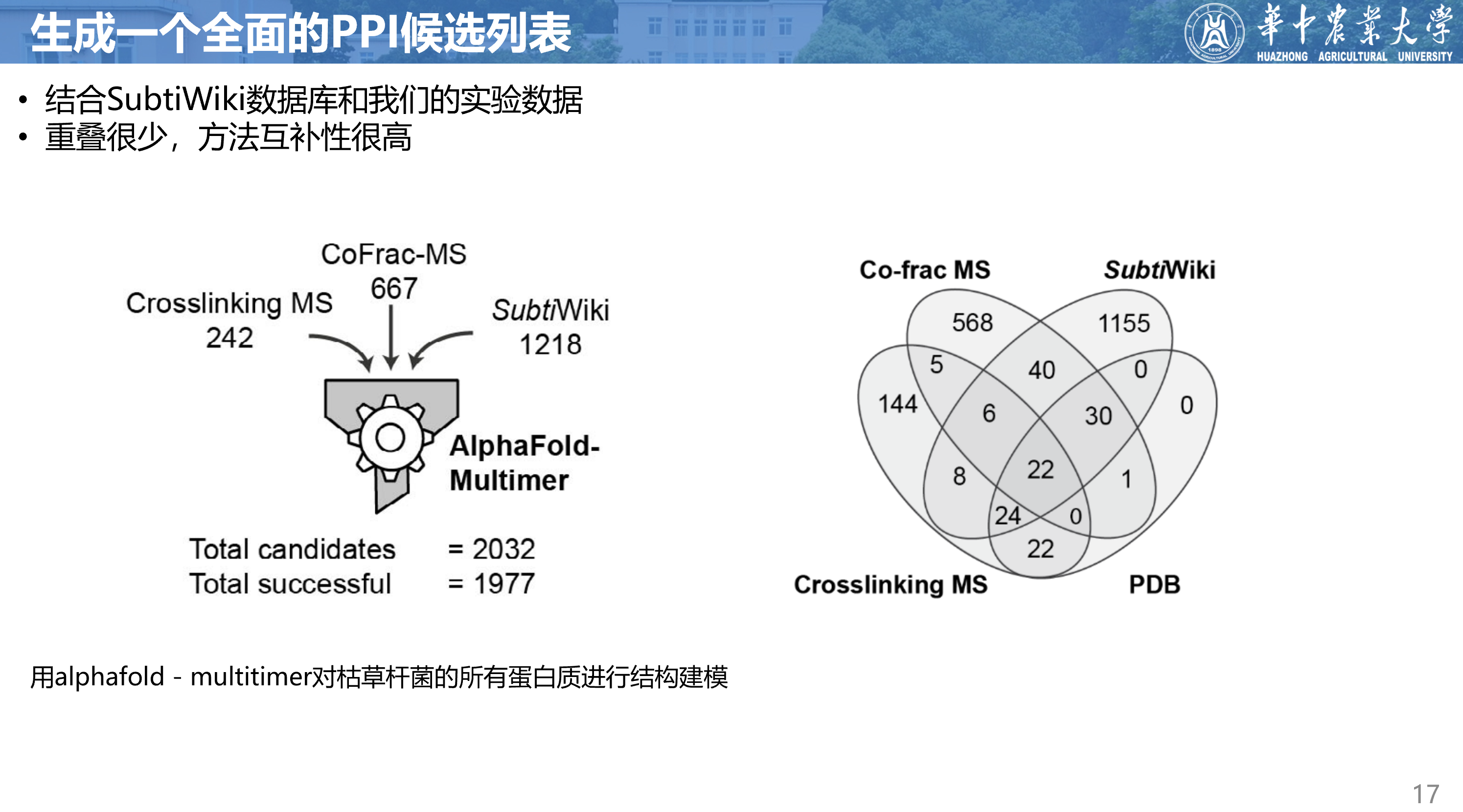
交联质谱和共分离质谱是互补的,它们把PPI和候选PPI的总数增加到了878,其中只有4%是重叠的。新发现的PPI已经被添加到了SubtiWiki数据库中。
SubtiWiki数据库是一个专门为模式生物枯草杆菌(Bacillus subtilis)建立的数据库,包含了该菌的基因组、代谢途径、相互作用、表达和调控等信息。

生成一个全面的PPI候选列表
为了得到一个全面的PPI候选列表,我们还把SubtiWiki数据库中已有的PPI加入了分析。这三个数据集(交联质谱、共分离质谱和数据库)之间的重叠很少(从右边的维恩图可以看到),这显示了不同方法的互补性。
用AlphaFold-Multimer识别蛋白质相互作用界面
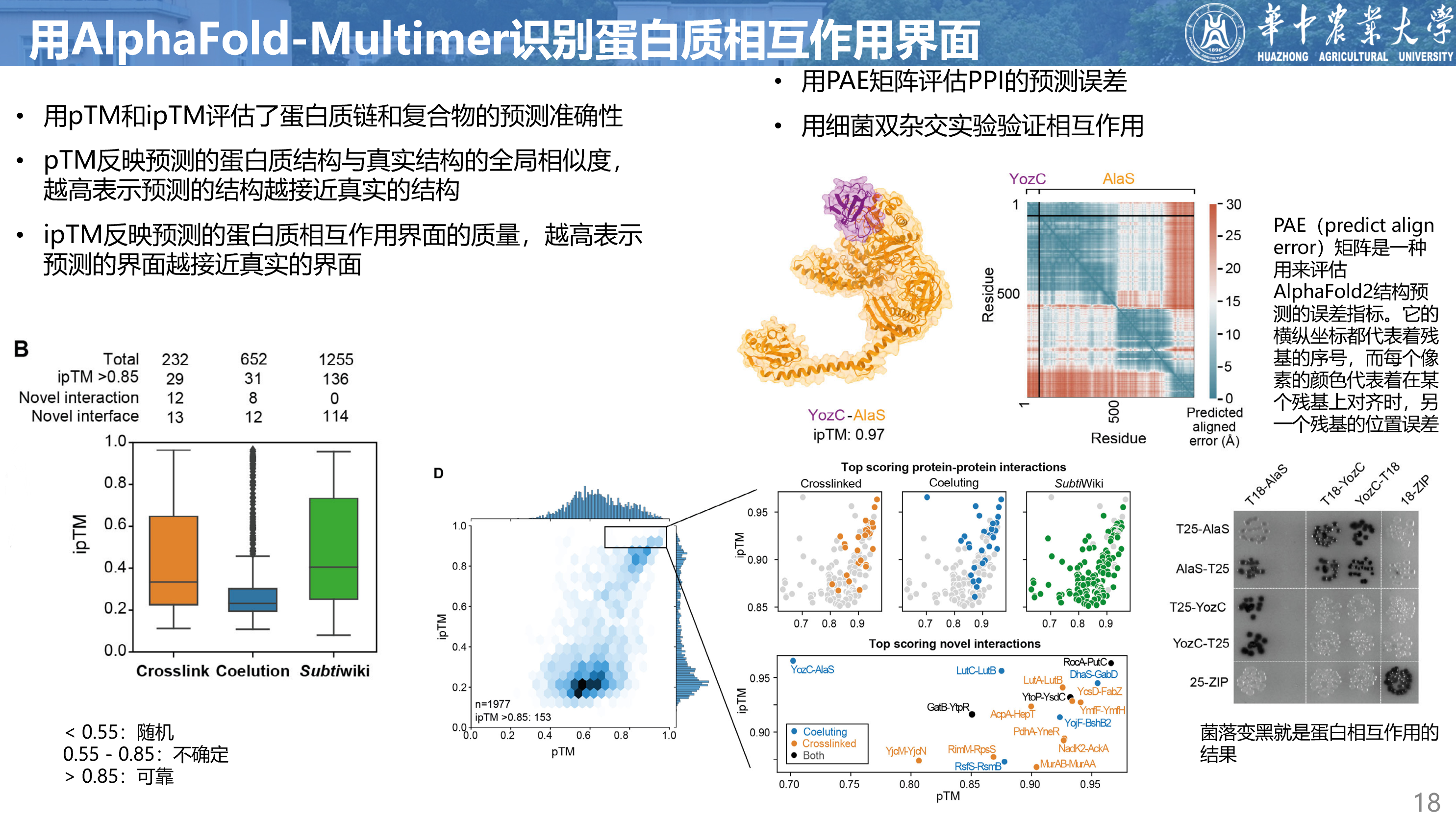
接下来,我们用alphafold预测了这些PPI复合物的结构,并用pTM和ipTM这两个指标评估了预测的质量。
pTM反映了预测的蛋白质结构与真实结构的全局相似度,ipTM反映了预测的蛋白质相互作用界面的质量。这两个值越高,表示预测得越好。
下图展示了三种方法(交联质谱、共分离质谱和数据库)产生的PPI的ipTM值的分布。
我们用ipTM = 0.85作为高置信度PPI模型的阈值。我们用随机的大肠杆菌蛋白对作为诱饵,发现AlphaFold-Multimer的ipTM < 0.55是随机的,0.55 - 0.85是不确定的,> 0.85是可靠的。我们建议对ipTM范围0.55-0.85的模型进行实验验证。
我们还用阈值筛选了三种方法产生的PPI,并比较了它们的分布。
此外,我们还用PAE(predicted aligned error)矩阵评估了每个预测的蛋白质相互作用的预测误差。PAE可以反映蛋白质或复合物的部分位置的置信度。图2E给出了一个例子,它显示了CoFrac-MS发现的AlaS和YozC之间的新型相互作用。这个模型的ipTM分数很高(0.97),但pTM分数较低(0.70),表示界面很可靠,但整体结构不确定。PAE图显示YozC和AlaS N端区域的相对位置预测误差很低,但这两个区域相对于AlaS的其他两个结构域的位置不确定。我们用细菌双杂交实验验证了这个相互作用。
细菌双杂交实验中,菌落变黑表示有相互作用发生。从图中可以看到,有几个菌落是黑色的,证实了AlaS和YozC之间存在相互作用。
到此为止,这篇文章发现PPI的主要流程就结束了。简单来说,就是先用交联质谱发现PPI,然后用共分离质谱发现PPI,再把数据库中已有的PPI加入分析,最后用alphafold预测它们的结构。
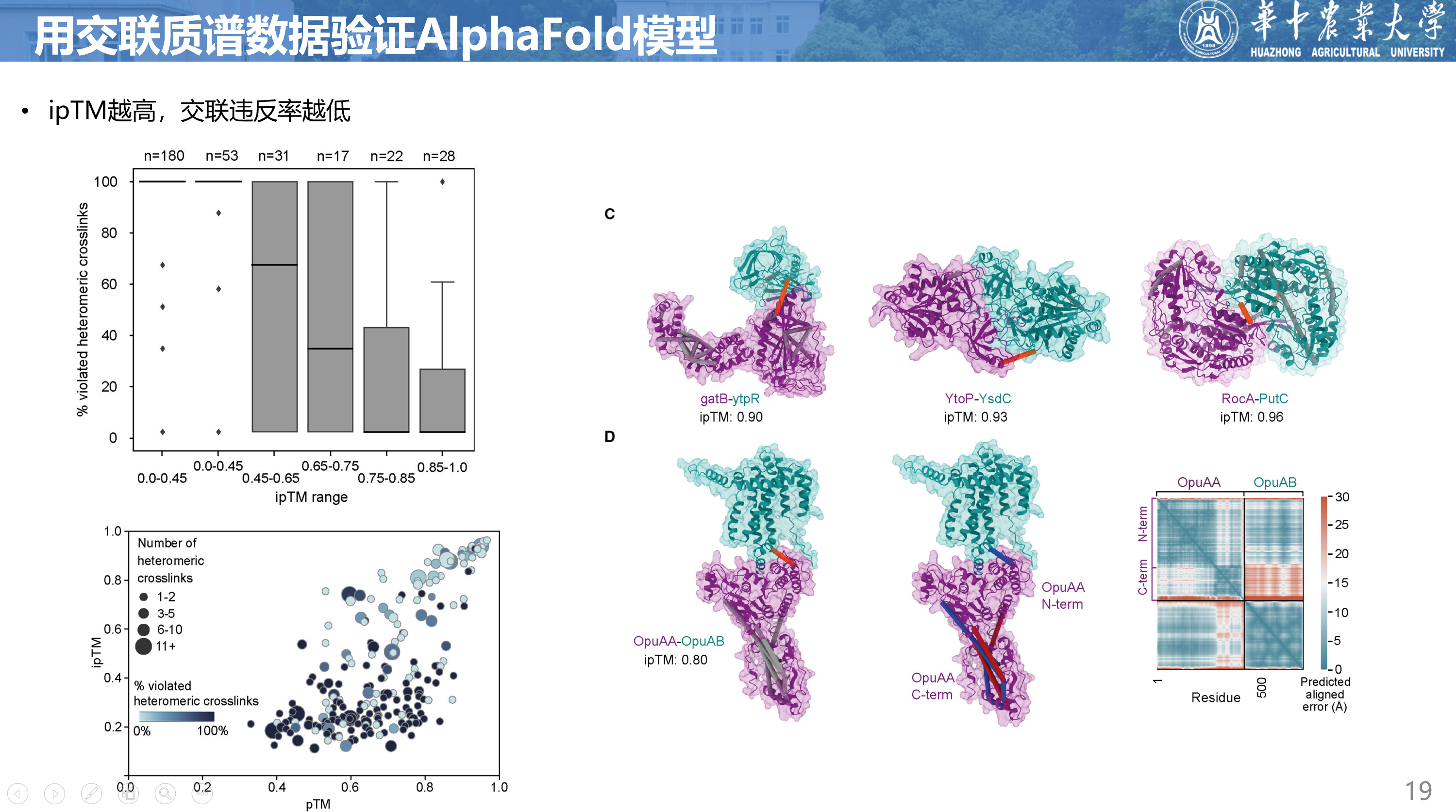
用交联质谱数据验证AlphaFold模型
接下来,作者用交联质谱数据验证了AlphaFold模型的准确性。
我们用交联质谱数据分析了细胞内蛋白质复合物的结构,并与相应的AlphaFold模型进行了比较(图3A)。我们发现ipTM和交联约束满足度呈现出很强的相关性,尽管AlphaFold模型预测时没有用到交联信息。当ipTM > 0.85时,交联违反率很低,表明高置信度的模型与细胞内观察到的残基间距一致。
从下图可以看出,ipTM > 0.85的模型的交联违反率很低。
在ipTM 0.55–0.85的范围内,模型的异源交联违反率分布很广,表明这些模型需要用实验数据进行验证或拒绝。交联违反率低说明预测的构象与细胞内的结构有一定的相似性。交联违反率高可能表示模型与细胞内的构象或真实解决方案不一致。此外,我们还发现,ipTM 0.75–0.85的模型比ipTM 0.55–75的模型更符合细胞内结构约束,表明模型质量随着ipTM分数的提高而提高。值得注意的是,ipTM低于0.55的模型的异源交联违反率中位数为100%,证实了低ipTM分数的模型界面质量差。
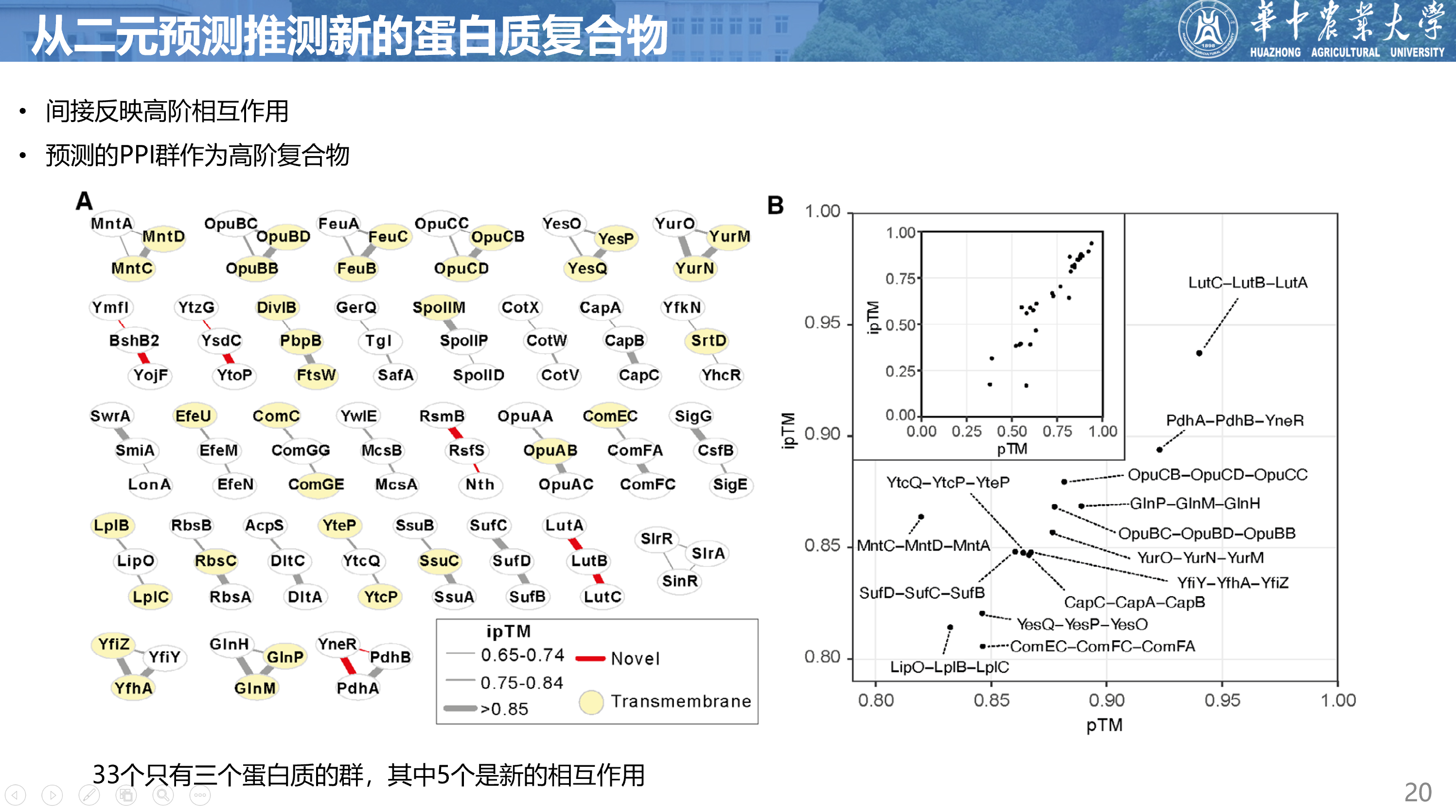
从二元预测推测新的蛋白质复合物
这部分的目的是通过PPI来找到多个蛋白质的复合体,这部分比较麻烦,因为这些PPI都是两个两个的,不能形成网络,不能利用从网络中挖掘社区的方法来找。
三种生成二元PPIs的方法(交联质谱、CoFrac-质谱和SubtiWiki数据库)受网络分析局限,只能间接反映高阶相互作用。上面预测的二元相互作用可能是独立的二元事件,也可能是更大的多蛋白复合物的一部分。这些复合物可能包含两种蛋白质的多个副本或涉及其他亚基。然而,二元相互作用可以用来推断更大组装体中的关联。
仔细说了几个蛋白复合体
我们用AlphaFold-Multimer(版本2.2.1;数据集EV6)预测了33个候选三聚体,得到了14个ipTM大于0.8的三聚体预测。其中排名第一的是一个未知的乳酸利用操纵子LutA-LutB-LutC(Chai等,2009)之间的复合物。这些相互作用是通过交联质谱(LutA-LutB)和CoFrac-MS(LutB-LutC)的组合确定的。在预测的结构中,PAE图显示了亚基全序列的高可信度的定位。LutB含有一个Fe-S簇,它位于亚基界面远处,但是LutC N端区域与覆盖Fe-S位点的LutB α2螺旋形成广泛的相互作用。
我们预测的复合物之一是CapA、CapB和CapC之间的复合物。这些蛋白质催化了细胞外聚合物γ-聚谷氨酸(PGA)的合成和出口。在枯草芽孢杆菌中,γ-PGA合成所需的所有酶都由capBCAE操纵子编码(Urushibata等,2002)。CapB和CapC形成γ-PGA合酶复合物,而CapA和CapE协同出口(Candela等,2005)。以前已经提出过CapBCA复合物的形成,其中连接酶亚基CapB和CapC之间有紧密的相互作用,而连接酶与CapA之间有松散的相互作用(Ashiuchi等,2001)。我们的工作为CapBCA复合物的存在提供了有力的结构预测证据(图4C)。有趣的是,在炭疽杆菌中,这是炭疽病的致病因子,cap操纵子存在于毒力质粒pXO2上。这种细菌利用γ-PGA囊来保护自己免受宿主免疫监视的攻击,使其成为一个重要的毒力因子(Mock & Fouet,2001;Jang等,2011)。
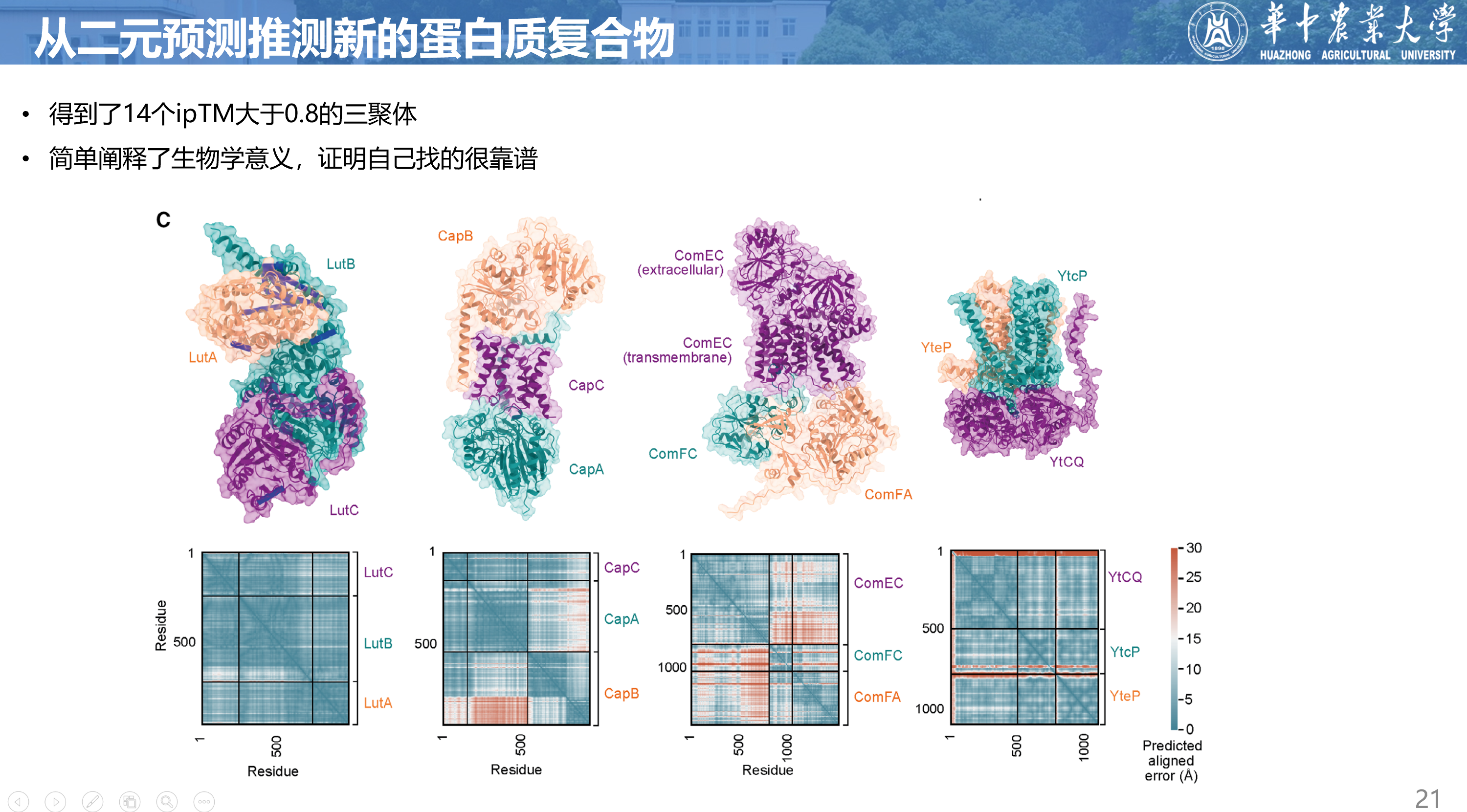
细分说了几个蛋白
后面作者就是仔细分析说了他们预测的一些关键复合物,我觉得这部分重要性不大。

总结
一句话总结就是:用交联质谱和共分离质谱识别PPI,并用AlphaFold-Multimer预测蛋白相互作用复合体的结构。