溶液环境中溶质离子或中间体的自由能计算是电化学研究中最棘手的问题之一。目前单纯的实验手段并不能对发生在溶液中的化学反应过程/机理进行直接探测,许多信息仍主要依赖于理论模拟。对于这一问题,目前很多研究者采用经验势场的溶剂模型方法,但其结果严重依赖于所使用的模型参数;而另外一种更为精确的热力学积分方法却相当昂贵,并且这种从头算方法并不总是适用于所有溶质物种。因此,目前还没有一种切实可行的从头算方法来对显式溶液环境中的基本热力学性质进行直接计算。
为解决这一问题,半导体所Lin-Wang Wang教授和天津大学杜希文教授、习聪博士等人开发了一种基于第一性原理分子动力学(AIMD)模拟的混合(显式+隐式)溶剂模型来精确计算离子在水中的溶解自由能。在该方法中,中心离子首先被两层显式水分子团簇包围,此水合离子外部再进一步应用隐式溶剂模型。每种结构进行15ps的AIMD计算,期间使用动态势阱来保持显式水分子一直包围在反应中心周围。最后用平均总能量作为反应的焓项,采用改进的原子自由度受力的准简谐近似用于计算熵贡献。最终计算得到的10种固体金属(Li、Na、K、Rb、Cs、Be、Mg、Ca、Sr、Ba)在水中的溶解电压和实验值相比的均方根误差在0.3 eV以内,并且对应的这10种气态离子在水中的溶解自由能的计算结果与实验值也非常接近。除自由能外,应用这种方法得到的水合阳离子的径向分布函数、阳离子周围第一层水团簇中的水分子个数等性质,均与实验结果一致,充分说明了此混合溶剂模型可以合理表征反应中心和水溶液之间的相互作用。
具体文章亮点有:
亮点1:巧妙设计的混合溶剂模型
混合溶剂模型如下图1所示,中心离子周围首先包围两层显示水分子以保证计算的精度,外围采用隐式溶剂模型以减小体系能量震荡同时降低计算成本。隐式溶剂模型的参数经过了精心调控以使显式-隐式溶剂模型界面无表面极矩。为了防止计算过程中显式水分子发散,采用了仅对水分子(氧原子)起作用的限域势能V(R)将水分子一直限制在距离结构中心Rcut半径范围内。同时结构采用恒定压力P和变化的Rcut使此混合溶剂模型可以合理应用在不同尺寸的体系中。因此,这一溶剂模型兼具精确性和普适性。
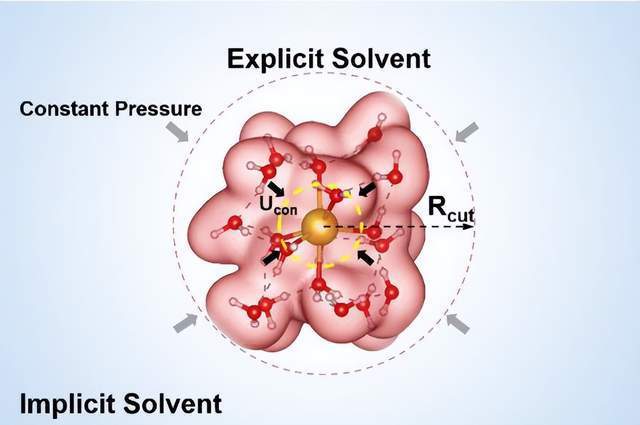
图1:混合溶剂模型示意图
亮点2:基于受力协方差矩阵的准简谐近似求得动力学中的收敛熵项
为求动力学中水分子的熵值,改进了传统的基于原子自由度的位移协方差矩阵求MD中体系熵值的方法,采用基于受力协方差矩阵的准简谐近似方法获得了随时间收敛的熵值。下图2中,对MD中水团簇和单个水分子熵值的求解中,原来位移方法求得的熵值会随模拟时长不断增大,而采用受力协方差矩阵求得的熵值会迅速收敛到一合理数值。
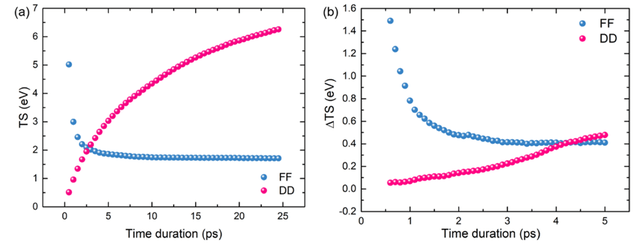
图2:(a) 显式水团簇的熵,(b)单个水分子的熵。分别使用位移协方差(DD)和受力协方差(FF)计算的平均值。DD方法存在熵值随模拟时长不收敛的问题。
亮点3:对不同价态阳离子溶解行为的精确模拟
采用本文中提出的混合溶剂模型进行AIMD计算的固体金属的溶解电压(图3)和实验值的均方根误差在0.3 eV以内。相较于其他方法(有限个显式水分子,或者隐式溶剂模型),混合溶剂模型的计算结果更准确;同时,采用混合溶剂模型计算的气态阳离子溶解自由能和已报道的其他理论方法对比(经验力场MD计算、Born模型等方法),本工作计算的样本最全面,和实验值相比的总体误差也更小。此外本工作还利用混合溶剂模型AIMD模拟计算了各个水合阳离子的半径、第一层水团簇中水分子的个数,和实验值也十分匹配。以上结果都充分说明了此溶剂模型方法可以合理精确表征反应中心和水分子之间的相互作用。
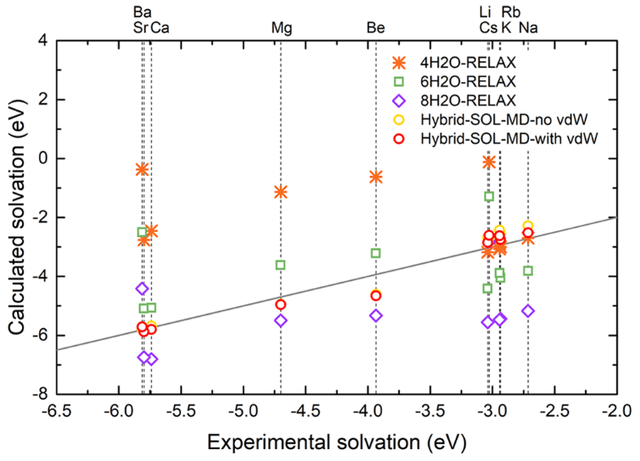
图3: 固体金属在水中的溶解电压的理论计算值和实验值对比。灰线表示二者完美匹配。相比于其他方法,采用本文中的混合溶剂模型计算出的溶解电压与实验值最接近。
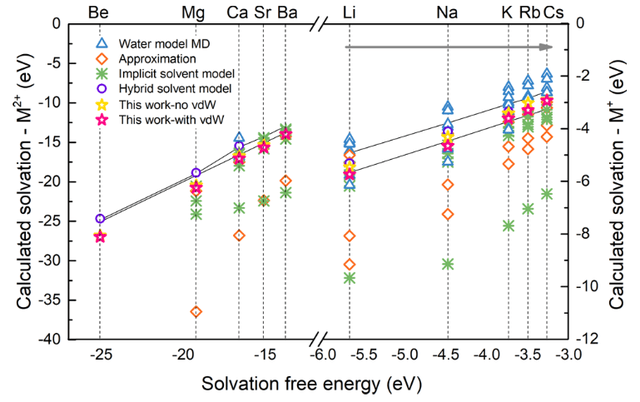
图4: 本文计算的气态离子溶解自由能与已报道的结果的比较。两条直线界定了实验值的范围。
总 结
溶液环境的正确模拟是探究很多化学现象的关键。它会影响分子的几何结构和电荷分布以及反应速率和热力学平衡等诸多性质。这项工作提出了一个精确并且切实可行的溶剂模型来合理表征实验中的溶液环境。这一方法适用于溶液环境中,或催化剂表面上的小分子溶质,无论它们是否带电。因此,该方法对研究电化学催化反应,以及局部溶剂效应具有重要意义。使用PWmat中的混合溶剂模型(Hybrid Solvent Model)结合固定电势法(Fix Potential)等特色功能,可以帮助我们快速探究真实的电极反应过程。
文献链接:
https://doi.org/10.1021/acs.jctc.1c01298
—E N D—
北京龙讯旷腾科技有限公司是成立于2015年的国家高新技术企业,是国内材料计算模拟工具软件研发创新的领导者,致力于开发满足“工业4.0”所需的原子精度材料研发Q-CAD(quantum-computer aided design)软件。公司自主开发的量子材料计算软件PWmat(平面波赝势方法并基于GPU加速)可以进行电子结构计算和从头算分子动力学模拟,适用于晶体、缺陷体系、半导体体系、金属体系、纳米体系、量子点、团簇和分子体系等。