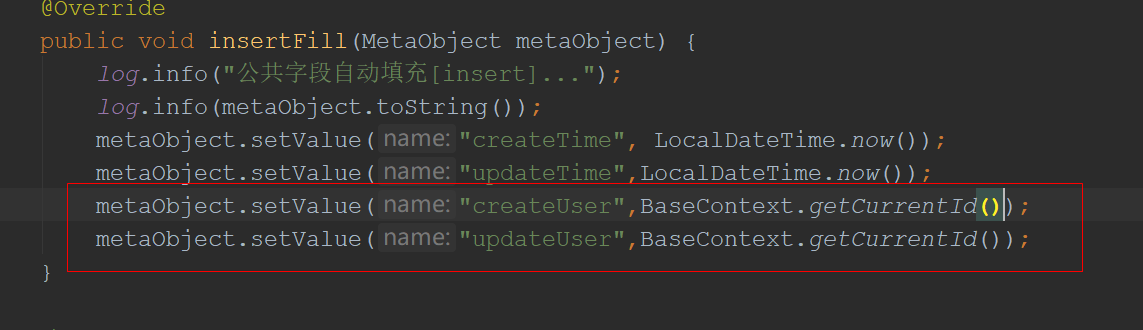
生成小分子力场TOP
ATB网站
生成基于Amber力场适配gromacs格式TOP文件,
对于使用GaussView 计算resp电荷可以参考: https://blog.csdn.net/weixin_42486623/article/details/129055384
下面我们使用上面是生成的mol2文件来生成基于Amber的力场,适配gromacs
- 检查缺失的参数
parmchk2 -i xx.mol2 -f mol2 -o xx.frcmod

2. 构建tleap.in文件
tleap.in如下:
source leaprc.gaff
MOL=loadmol2 methane.mol2
loadamberparams methane.frcmod
saveamberparm MOL MOL.prmtop MOL.rst7
savepdb MOL MOL_tleap.pdb
quit
执行tleap.in文件
tleap -f ./tleap.in
生成两个文件



3. 使用acpype 转化为gromacs的Top文件
acpype -p MOL.prmtop -x MOL.rst7
使用mktop_2.2.1.pl生成OPLS力场
- 下载mktop_2.2.1.pl文件,http://www.aribeiro.net.br/mktop/
- 用vi编辑器打开mktop_2.2.1.pl,修改gromacs的力场目录为自己linux系统安装gromacs的路径,即修改如下红色框里面的内容。(或者在win系统下面先修改好之后再上传上去)

- 创建好自己分子的pdb文件, 可以使用GaussView 6.0软件画分子结构图
- 由于此方法不能生成电荷,所以需要自己准备(可使用高斯等量化软件自己优化生成)按照pdb文件里面的原子顺序准备一个电荷文件(取任意名,比如:charge.txt)。电荷文件的内容仿照如下甲烷的格式编写: (切记,顺序一定要和pdb文件里面的原子顺序一致) 具体方法可以参考:https://blog.csdn.net/weixin_42486623/article/details/129055384

再次核对一下是不是和甲烷的pdb原子对应:

- 把pdb文件转成linux格式的,运行下面代码:https://blog.csdn.net/weixin_42486623/article/details/129055384
dos2unix **.pdb
- 执行命令:
perl mktop_2.2.1.pl -i methane.pdb -c charge.txt -o top.top -ff opls -conect yes
- 若已经有top文件,仅仅是需要生成这个单分子的itp,那么可以修改生成的top.top文件名字为:**.itp,并在已有的top文件里面include即可。但是还需要删除top.top里面的一些内容,需要删除的内容如下图红框所示:


使用LigParGen在线工具生成OPLS力场
地址: http://zarbi.chem.yale.edu/ligpargen/index.html