之前笔者介绍过一步法的分析的流程: WGCNA加权基因共表达网络一步法分析学习 https://mp.weixin.qq.com/s/2Q37RcJ1pBy_WO1Es8upIg
建议先看一下之前的推文,了解一下WGCNA的基础原理。
这次就来介绍一下多步法
分析步骤:
1.导入
rm(list=ls())
library(WGCNA)
library(ggplot2)
load("data.Rdata")
exp <- exprSet
pd <- meta
2.数据预处理
如果有批次效应,需要先进行去除;
如果数据存在系统偏移,需要进行quantile normalization;
标准化推荐使用DESeq2中的varianceStabilizingTransformation方法,
或将基因标准化后的数据(如FPKM、CPM等)进行log2(x+1)转化。
# 大家可以依据自己数据的实际情况进行这些步骤
identical(colnames(exp),rownames(pd))
boxplot(exp)
3.提取表达矩阵并进行质控
datExpr0 = t(exp[order(apply(exp, 1, mad), decreasing = T)[1:5000],])
#datExpr0 = t(exp[order(apply(exp, 1, var), decreasing = T)[1:round(0.25*nrow(exp))],])
#datExpr0 = as.data.frame(t(exp))
datExpr0[1:4,1:4]
#rownames(datExpr0) = names(exp)[-c(1:8)]
# 基因和样本质控----
gsg = goodSamplesGenes(datExpr0, verbose = 3)
# `verbose` 参数被设置为 `3`,用于控制函数 `goodSamplesGenes` 的详细输出程度。
# `verbose = 0`:不产生任何输出,只返回结果,通常用于静默模式。
# `verbose = 1`:产生基本的信息输出,以提供一些关于函数执行进度的信息。
# `verbose = 2`:产生更详细的输出,可能包括一些中间步骤的信息。
# `verbose = 3`:产生最详细的输出,通常包括每个步骤的详细信息,用于调试和详细分析。
gsg$allOK # 返回TRUE则继续
if (!gsg$allOK){
# 把含有缺失值的基因或样本打印出来
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# 去掉那些缺失值
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
# 样本质控
sampleTree = hclust(dist(datExpr0), method = "average")
# 使用欧氏距离 (dist(dat)) 计算样本间的距离矩阵。
# 使用层次聚类方法(平均连接法)构建聚类树。
png(file = "2.sampleClustering.png", width = 10000, height = 2000,res = 300)
par(cex = 0.6)
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers",
sub="", xlab="", cex.lab = 1.5,cex.axis = 1.5, cex.main = 2)
#abline(h = 140, col = "red") #根据实际情况而定是否要划线去除离群点
dev.off()
4.异常值筛选及质控
# 如果有异常值就需要去掉
# if(T){
# clust = cutreeStatic(sampleTree, cutHeight = 150, minSize = 10)
# table(clust) # 0代表切除的,1代表保留的
# keepSamples = (clust!=0)
# datExpr = datExpr0[keepSamples, ]
# }
# cutHeight = 200:用于指定在层次聚类树中切割的高度。在树状结构中,高度表示样本之间的相似性或距离。
# 通过指定 cutHeight,你可以控制在哪个高度水平切割树,从而确定最终的簇数。
# minSize = 10:用于指定最小簇的大小。在进行切割时,如果某个簇的大小小于 minSize,
# 则可能会合并到其他簇中,以确保生成的簇都具有足够的样本数。
# 切除完了之后需要再回到上面的代码进行做图!
# #去除了异常样本之后需要跟临床信息做一个对应哦
# s <- intersect(rownames(datExpr),rownames(pd))
# pd <- pd[s,]
# datExpr <- datExpr[s,]
# a <- identical(rownames(datExpr),rownames(pd));a
#没有异常样本就不需要去除
datExpr = datExpr0
5.添加表型矩阵联合
# 这里要注意的是,用于关联分析的性状必须是数值型特征(比如年龄、体重、基因表达水平等)。
# 如果是分类变量(比如性别、地区等),则需要转换为0-1矩阵的形式
# (1表示属于此组或有此属性,0表示不属于此组或无此属性)。
library(stringr)
#搜索列名
tmp = data.frame(colnames(pd))
traitData = data.frame(
Female = as.numeric(grepl("^FEMALE$", pd$gender)),
Male = as.numeric(grepl("^MALE$", pd$gender))
)
str(traitData)
table(traitData[,1])
dim(traitData)
names(traitData)
datTraits = traitData
#表型和样本的相关性----
sampleTree2 = hclust(dist(datExpr), method = "average")
# 用颜色表示表型在各个样本的表现: 白色表示低,红色为高,灰色为缺失
traitColors = numbers2colors(datTraits, signed = FALSE)
# 把样本聚类和表型热图绘制在一起
png(file = "3.Sample dendrogram and trait heatmap.png",
width = 10000, height = 2000,res = 300)
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
dev.off()

6.软阈值筛选
#设置一系列软阈值(默认1到30)
powers = c(1:10, seq(from = 12, to=30, by=2))
#帮助用户选择合适的软阈值,进行拓扑网络分析
#需要输入表达矩阵、设置的阈值范围、运行显示的信息程度(verbose=0不显示任何信息)
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
sft$powerEstimate
# pickSoftThreshold 函数的输出,用于选择最佳的软阈值以构建基因共表达网络或网络模型。
# powerEstimate:这是估计的最佳软阈值的值,它是一个整数。在这个结果中,估计的最佳软阈值是4。
# fitIndices:这是一个数据框,包含了不同软阈值下的拟合指标。每一行代表一个不同的软阈值(在 powers 中定义),列包括以下信息:
# Power:软阈值的幂次。
# SFT.R.sq:Scale Free Topology Model Fit,表示拟合模型的拟合度,通常用 R^2 衡量。较高的值表示模型更好地拟合数据。
# slope:表示软阈值幂次与拟合模型的斜率,通常用于衡量网络的无标度拓扑结构。一般来说,斜率绝对值越小,模型越能保持无标度拓扑结构。
# truncated.R.sq:表示截断模型的拟合度,也是一个衡量模型拟合程度的指标。
# mean.k.:平均连接度(Mean Connectivity),用于描述网络中节点的连接性。
# median.k.:中位数连接度,是平均连接度的中位数。
# max.k.:最大连接度,表示网络中具有最多连接的节点的连接数。
png(file = "4.Soft threshold.png", width = 2000, height = 1500,res = 300)
par(mfrow = c(1,2))
cex1 = 0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",
ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"))
#注意这里的-sign(sft$fitIndices[,3])中sign函数,它把正数、负数分别转为1、-1
text(sft$fitIndices[,1],
-sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
# 设置筛选标准h=r^2^=0.9。这里的0.9是个大概的数,就是看左图软阈值6大概对应的位置
abline(h=cex1,col="red")
#看一下Soft threshold与平均连通性
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",
ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
dev.off()

6.1经验power (无满足条件的power时选用)
# 无向网络在power小于15或有向网络power小于30内,没有一个power值可以使
# 无标度网络图谱结构R^2达到0.8,平均连接度较高如在100以上,可能是由于
# 部分样品与其他样品差别太大。这可能由批次效应、样品异质性或实验条件对表达影响太大等造成。可以通过绘制样品聚类查看分组信息和有无异常样品。
# 如果这确实是由有意义的生物变化引起的,也可以使用下面的经验power值。
if (is.na(sft$powerEstimate)){
power = ifelse(nSamples<20, ifelse(type == "unsigned", 9, 18),
ifelse(nSamples<30,
ifelse(type == "unsigned", 8, 16),
ifelse(nSamples<40, ifelse(type == "unsigned", 7, 14),
ifelse(type == "unsigned", 6, 12))
)
) }
7.网络构建
多步法构建网络
## 构建加权共表达网络分为两步:
## 1. 计算邻近值,也是就是两个基因在不同样品中表达量的表达相关系数(pearson correlation rho),
## 2. 计算topology overlap similarity (TOM)。用TOM表示两个基因在网络结构上的相似性,即两个基因如果具有相似的邻近基因,这两个基因更倾向于有相互作用。
str(datExpr)
#datExpr <- as.data.frame(lapply(datExpr, as.numeric))
sft$powerEstimate
softPower <- 4 #根据前面的结果进行更改
###(1)网络构建 Co-expression similarity and adjacency
adjacency = adjacency(datExpr, power = softPower)
###(2) 邻近矩阵到拓扑矩阵的转换,Turn adjacency into topological overlap
TOM = TOMsimilarity(adjacency)
dissTOM = 1-TOM
###(3) 聚类拓扑矩阵 Clustering using TOM
# Call the hierarchical clustering function
geneTree = hclust(as.dist(dissTOM), method = "average");
# Plot the resulting clustering tree (dendrogram)
## 这个时候的geneTree与一步法的 net$dendrograms[[1]] 性质类似,但是还需要进行进一步处理
pdf("geneTree_plot.pdf", width = 10, height = 7) # 或者使用 png() 等函数
plot(geneTree, xlab="", sub="", main = "Gene clustering on TOM-based dissimilarity",
labels = FALSE, hang = 0.04)
dev.off() # 关闭图形设备
###(4) 聚类分支的修整 dynamicTreeCut
################# set the minimum module size ##############################
minModuleSize = 30
####
# Module identification using dynamic tree cut:
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM,
deepSplit = 2, pamRespectsDendro = FALSE,
minClusterSize = minModuleSize)
table(dynamicMods)
# Convert numeric lables into colors
dynamicColors = labels2colors(dynamicMods)
table(dynamicColors)
# Plot the dendrogram and colors underneath
plotDendroAndColors(geneTree, dynamicColors, "Dynamic Tree Cut",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05,
main = "Gene dendrogram and module colors")
###(5) 聚类结果相似模块的融合 Merging of modules whose expression profiles are very similar
# Calculate eigengenes
MEList = moduleEigengenes(datExpr, colors = dynamicColors)
MEs = MEList$eigengenes
# Calculate dissimilarity of module eigengenes
MEDiss = 1-cor(MEs)
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average")
#一般选择 height cut 为0.25,对应于有75%相关性,进行融合
###################### set Merging height cut ################################
MEDissThres = 0.5
####
# Plot the result
plot(METree, main = "Clustering of module eigengenes",
xlab = "", sub = "")
# Plot the cut line into the dendrogram
abline(h=MEDissThres, col = "red")
# Call an automatic merging function
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)
# The merged module colors
mergedColors = merge$colors
# Eigengenes of the new merged modules:
mergedMEs = merge$newMEs
# 统计mergedmodule
table(mergedColors)
### (6) plot the gene dendrogram
pdf(file = "step3_stepbystep_DynamicTreeCut_genes-modules.pdf", width = 16,height = 12)
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors),
c("Dynamic Tree Cut", "Merged dynamic"),
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()
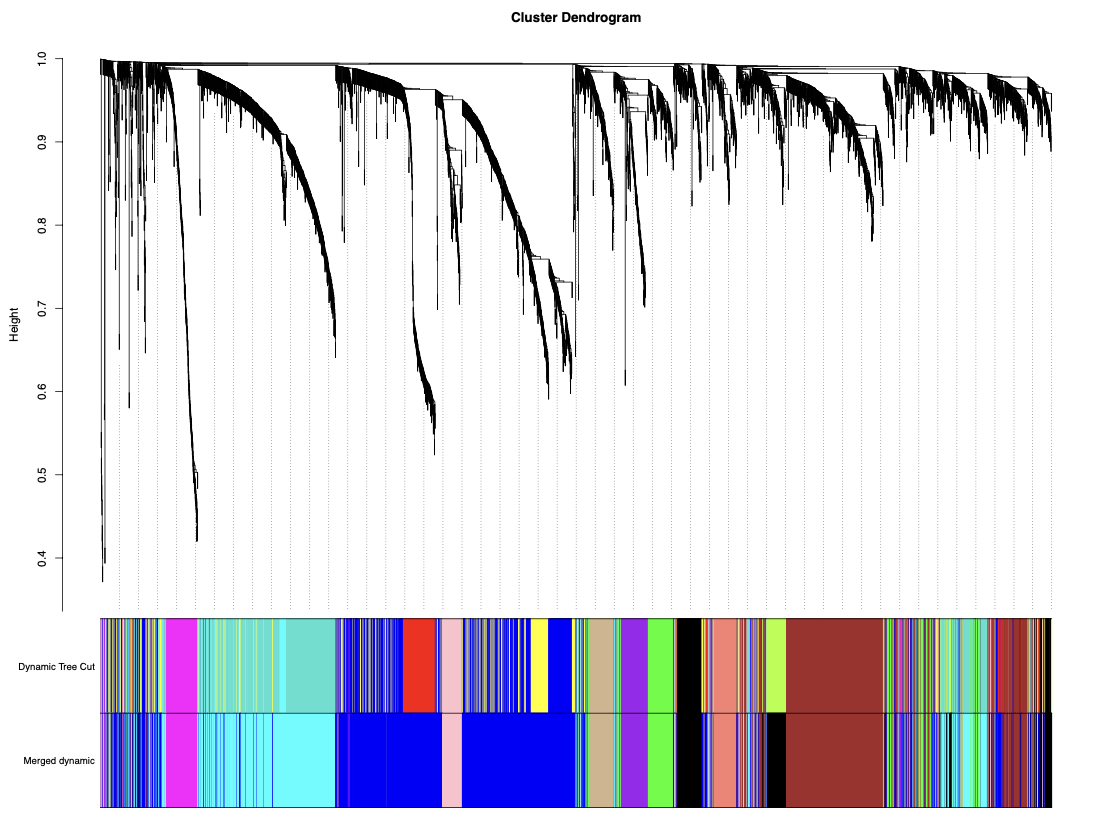
8.保存数据
# Rename to moduleColors
moduleColors = mergedColors
# Construct numerical labels corresponding to the colors
colorOrder = c("grey", standardColors(50))
moduleLabels = match(moduleColors, colorOrder)-1
MEs = mergedMEs
# Save module colors and labels for use in subsequent parts
save(MEs, moduleLabels, moduleColors, geneTree,
file = "step3_stepByStep_genes_modules.Rdata")
nrow(datExpr) # 查看 datExpr 的行数(基因数)
length(moduleColors)
geneNames = rownames(t(datExpr))
# 合并后的模块颜色
moduleColors = mergedColors
# 创建一个数据框,包含基因名称和它们对应的模块颜色
geneModule = data.frame(Gene = geneNames, Module = moduleColors)
# 查看每个模块的基因数量
table(geneModule$Module)
# 例如,提取某个特定模块(例如 "blue" 模块)中的基因:
blueModuleGenes = geneModule$Gene[geneModule$Module == "blue"]
# 查看蓝色模块中的基因
print(blueModuleGenes)
# 保存到 CSV 文件
write.csv(geneModule, file = "Module_Genes.csv", row.names = FALSE)
最关键的就是获得合并后的模块中的基因啦~
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -