介绍
RNA-seq 目前是测量细胞反应的最突出的方法之一。RNA-seq 不仅能够分析样本之间基因表达的差异,还可以发现新的亚型并分析 SNP 变异。本教程[1]将涵盖处理和分析差异基因表达数据的基本工作流程,旨在提供设置环境和运行比对工具的通用方法。由于完整版过长,因此分为两部分,需要获取完整版的,请跳转文末。
7. 差异分析
-
将基因计数导入 R/RStudio
工作流程完成后,您现在可以使用基因计数表作为 DESeq2 的输入,使用 R 语言进行统计分析。
7.1. 安装R包
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2") ; library(DESeq2)
biocLite("ggplot2") ; library(ggplot2)
biocLite("clusterProfiler") ; library(clusterProfiler)
biocLite("biomaRt") ; library(biomaRt)
biocLite("ReactomePA") ; library(ReactomePA)
biocLite("DOSE") ; library(DOSE)
biocLite("KEGG.db") ; library(KEGG.db)
biocLite("org.Mm.eg.db") ; library(org.Mm.eg.db)
biocLite("org.Hs.eg.db") ; library(org.Hs.eg.db)
biocLite("pheatmap") ; library(pheatmap)
biocLite("genefilter") ; library(genefilter)
biocLite("RColorBrewer") ; library(RColorBrewer)
biocLite("GO.db") ; library(GO.db)
biocLite("topGO") ; library(topGO)
biocLite("dplyr") ; library(dplyr)
biocLite("gage") ; library(gage)
biocLite("ggsci") ; library(ggsci)
7.2. 导入表达矩阵
-
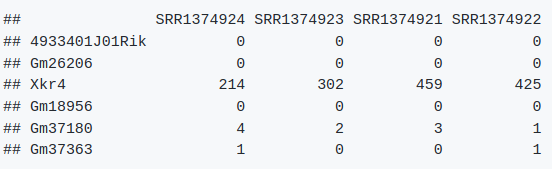
开始导入文件夹中的 featureCounts表。本教程将使用DESeq2对样本组之间进行归一化和执行统计分析。
# 导入基因计数表
# 使行名成为基因标识符
countdata <- read.table("example/final_counts.txt", header = TRUE, skip = 1, row.names = 1)
# 从列标识符中删除 .bam 和 '..'
colnames(countdata) <- gsub(".bam", "", colnames(countdata), fixed = T)
colnames(countdata) <- gsub(".bam", "", colnames(countdata), fixed = T)
colnames(countdata) <- gsub("..", "", colnames(countdata), fixed = T)
# 删除长度字符列
countdata <- countdata[ ,c(-1:-5)]
# 查看 ID
head(countdata) # 如下图
7.3. 导入metadata
-
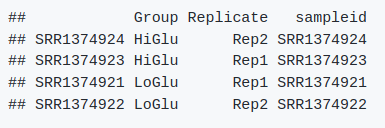
导入元数据文本文件。 SampleID必须是第一列。
# 导入元数据文件
# 使行名称与 countdata 中的 sampleID 相匹配
metadata <- read.delim("example/metadata.txt", row.names = 1)
# 将 sampleID 添加到映射文件
metadata$sampleid <- row.names(metadata)
# 重新排序 sampleID 以匹配 featureCounts 列顺序。
metadata <- metadata[match(colnames(countdata), metadata$sampleid), ]
# 查看 ID
head(metadata) # 如下图
7.4. DESeq2对象
-
根据计数和元数据创建 DESeq2对象
# - countData : 基于表达矩阵
# - colData : 见上图
# - design : 比较
ddsMat <- DESeqDataSetFromMatrix(countData = countdata,
colData = metadata,
design = ~Group)
# 查找差异表达基因
ddsMat <- DESeq(ddsMat)
7.5. 统计
-
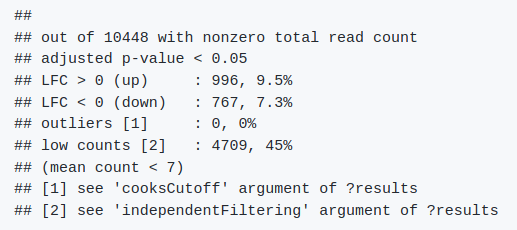
获取基因数量的基本统计数据
# 使用 FDR 调整 p-values 从检测中获取结果
results <- results(ddsMat, pAdjustMethod = "fdr", alpha = 0.05)
# 结果查看
summary(results) # 如下图
# 检查 log2 fold change
## Log2 fold change is set as (LoGlu / HiGlu)
## Postive fold changes = Increased in LoGlu
## Negative fold changes = Decreased in LoGlu
mcols(results, use.names = T) # 结果如下

8. 注释基因symbol
经过比对和总结,我们只有带注释的基因符号。要获得有关基因的更多信息,我们可以使用带注释的数据库将基因符号转换为完整的基因名称和 entrez ID 以进行进一步分析。
-
收集基因注释信息
# 小鼠基因组数据库
library(org.Mm.eg.db)
# 添加基因全名
results$description <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "GENENAME",
keytype = "SYMBOL",
multiVals = "first")
# 添加基因 symbol
results$symbol <- row.names(results)
# 添加 ENTREZ ID
results$entrez <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "ENTREZID",
keytype = "SYMBOL",
multiVals = "first")
# 添加 ENSEMBL
results$ensembl <- mapIds(x = org.Mm.eg.db,
keys = row.names(results),
column = "ENSEMBL",
keytype = "SYMBOL",
multiVals = "first")
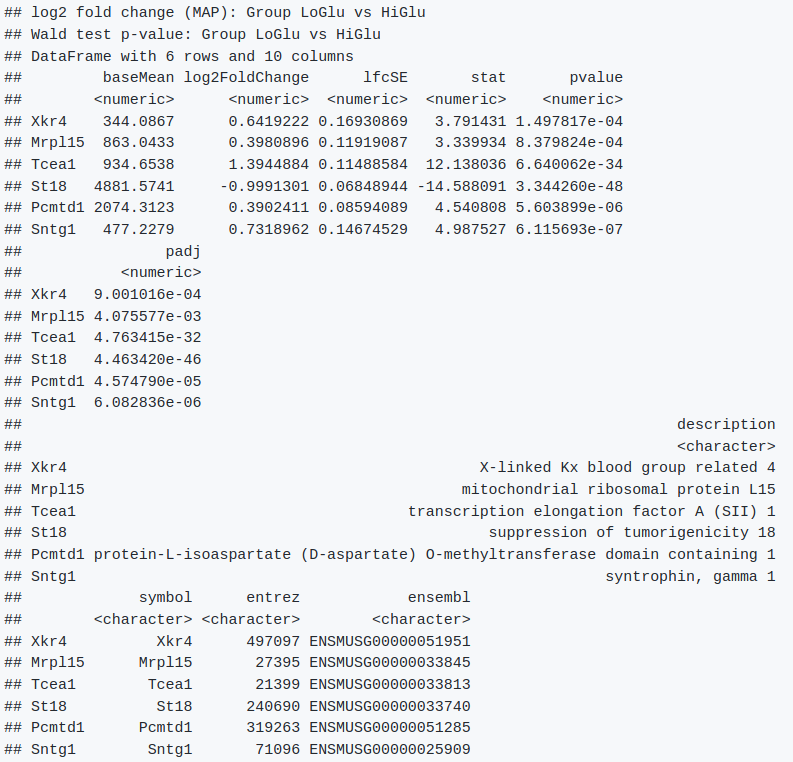
# 取 (q < 0.05) 的基因
results_sig <- subset(results, padj < 0.05)
# 查看结果
head(results_sig) # 如下图
-
将所有重要结果写入 .txt 文件
# 将归一化基因计数写入 .txt 文件
write.table(x = as.data.frame(counts(ddsMat), normalized = T),
file = 'normalized_counts.txt',
sep = '\t',
quote = F,
col.names = NA)
# 将标准化基因计数写入 .txt 文件
write.table(x = counts(ddsMat[row.names(results_sig)], normalized = T),
file = 'normalized_counts_significant.txt',
sep = '\t',
quote = F,
col.names = NA)
# 将带注释的结果表写入 .txt 文件
write.table(x = as.data.frame(results),
file = "results_gene_annotated.txt",
sep = '\t',
quote = F,
col.names = NA)
# 将重要的注释结果表写入 .txt 文件
write.table(x = as.data.frame(results_sig),
file = "results_gene_annotated_significant.txt",
sep = '\t',
quote = F,
col.names = NA)
9. 绘图
有多种方法可以绘制基因表达数据。下面只列出了一些流行的方法。
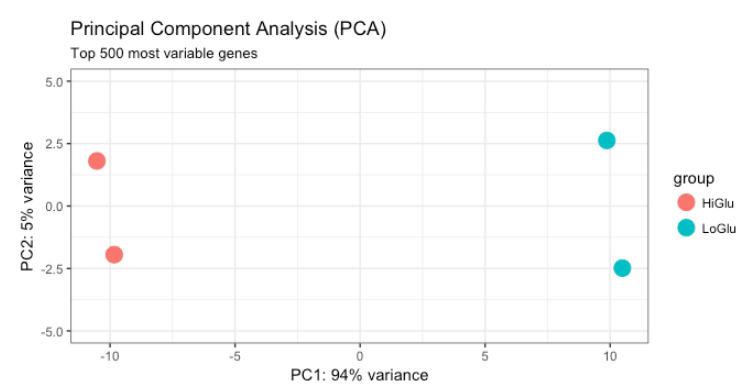
9.1. PCA
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 按列变量绘制 PCA
plotPCA(ddsMat_rlog, intgroup = "Group", ntop = 500) +
theme_bw() +
geom_point(size = 5) +
scale_y_continuous(limits = c(-5, 5)) +
ggtitle(label = "Principal Component Analysis (PCA)",
subtitle = "Top 500 most variable genes")
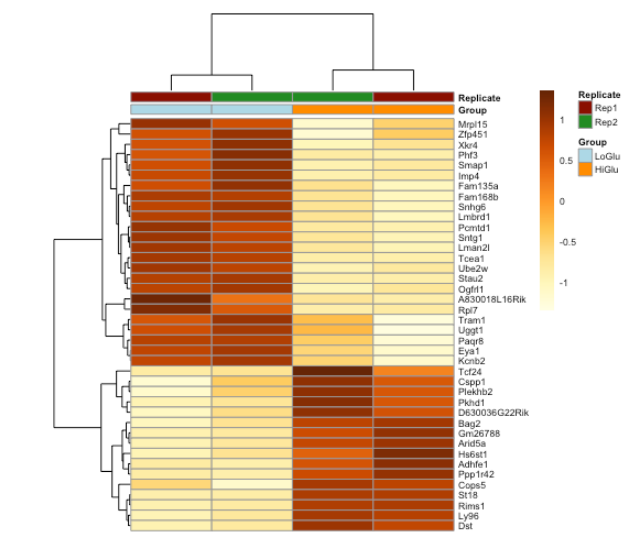
9.2. Heatmap
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 收集30个显著基因,制作矩阵
mat <- assay(ddsMat_rlog[row.names(results_sig)])[1:40, ]
# 选择您要用来注释列的列变量。
annotation_col = data.frame(
Group = factor(colData(ddsMat_rlog)$Group),
Replicate = factor(colData(ddsMat_rlog)$Replicate),
row.names = colData(ddsMat_rlog)$sampleid
)
# 指定要用来注释列的颜色。
ann_colors = list(
Group = c(LoGlu = "lightblue", HiGlu = "darkorange"),
Replicate = c(Rep1 = "darkred", Rep2 = "forestgreen")
)
# 使用 pheatmap 功能制作热图。
pheatmap(mat = mat,
color = colorRampPalette(brewer.pal(9, "YlOrBr"))(255),
scale = "row",
annotation_col = annotation_col,
annotation_colors = ann_colors,
fontsize = 6.5,
cellwidth = 55,
show_colnames = F)
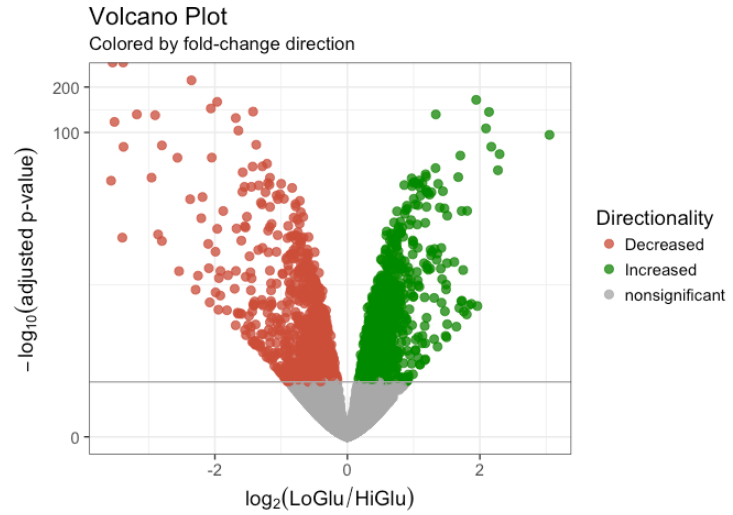
9.3. Volcano
# 从 DESeq2 结果中收集倍数变化和 FDR 校正的 pvalue
## - 将 pvalues 更改为 -log10 (1.3 = 0.05)
data <- data.frame(gene = row.names(results),
pval = -log10(results$padj),
lfc = results$log2FoldChange)
# 删除任何以 NA 的行
data <- na.omit(data)
## If fold-change > 0 and pvalue > 1.3 (Increased significant)
## If fold-change < 0 and pvalue > 1.3 (Decreased significant)
data <- mutate(data, color = case_when(data$lfc > 0 & data$pval > 1.3 ~ "Increased",
data$lfc < 0 & data$pval > 1.3 ~ "Decreased",
data$pval < 1.3 ~ "nonsignificant"))
# 用 x-y 值制作一个基本的 ggplot2 对象
vol <- ggplot(data, aes(x = lfc, y = pval, color = color))
# 添加 ggplot2 图层
vol +
ggtitle(label = "Volcano Plot", subtitle = "Colored by fold-change direction") +
geom_point(size = 2.5, alpha = 0.8, na.rm = T) +
scale_color_manual(name = "Directionality",
values = c(Increased = "#008B00", Decreased = "#CD4F39", nonsignificant = "darkgray")) +
theme_bw(base_size = 14) +
theme(legend.position = "right") +
xlab(expression(log[2]("LoGlu" / "HiGlu"))) +
ylab(expression(-log[10]("adjusted p-value"))) +
geom_hline(yintercept = 1.3, colour = "darkgrey") +
scale_y_continuous(trans = "log1p")
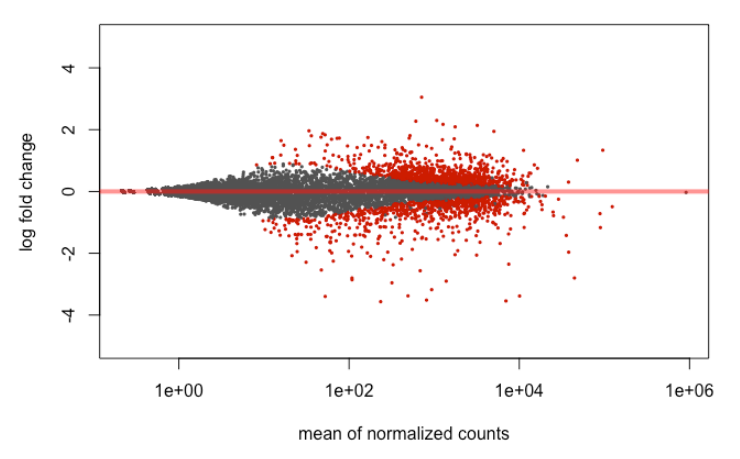
9.4. MA
plotMA(results, ylim = c(-5, 5))
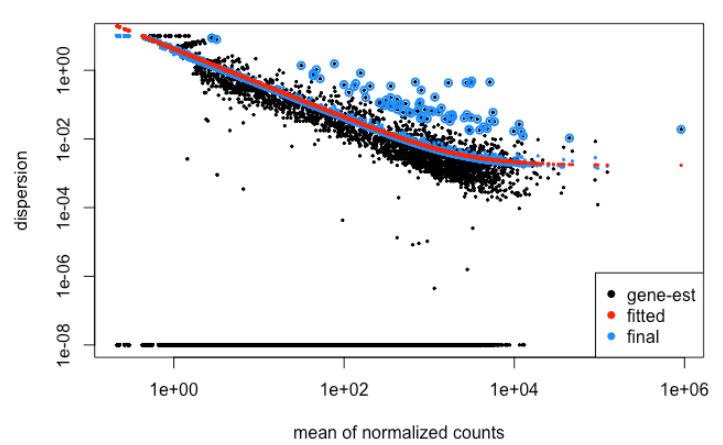
9.5. Dispersions
plotDispEsts(ddsMat)
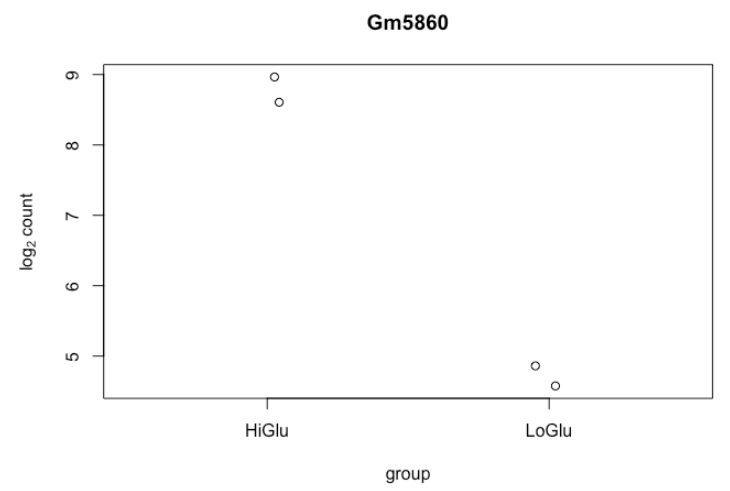
9.6. 单基因图
# 将所有样本转换为 rlog
ddsMat_rlog <- rlog(ddsMat, blind = FALSE)
# 获得最高表达的基因
top_gene <- rownames(results)[which.min(results$log2FoldChange)]
# 画单基因图
plotCounts(dds = ddsMat,
gene = top_gene,
intgroup = "Group",
normalized = T,
transform = T)
10. 通路富集
-
从差异表达基因中寻找通路
通路富集分析是基于单个基因变化生成结论的好方法。有时个体基因的变化是难以解释。但是通过分析基因的通路,我们可以收集基因反应的视图。
设置矩阵以考虑每个基因的 EntrezID 和倍数变化
# 删除没有任何 entrez 标识符的基因
results_sig_entrez <- subset(results_sig, is.na(entrez) == FALSE)
# 创建一个log2倍数变化的基因矩阵
gene_matrix <- results_sig_entrez$log2FoldChange
# 添加 entrezID 作为每个 logFC 条目的名称
names(gene_matrix) <- results_sig_entrez$entrez
# 查看基因矩阵的格式
##- Names = ENTREZ ID
##- Values = Log2 Fold changes
head(gene_matrix) # 如下图

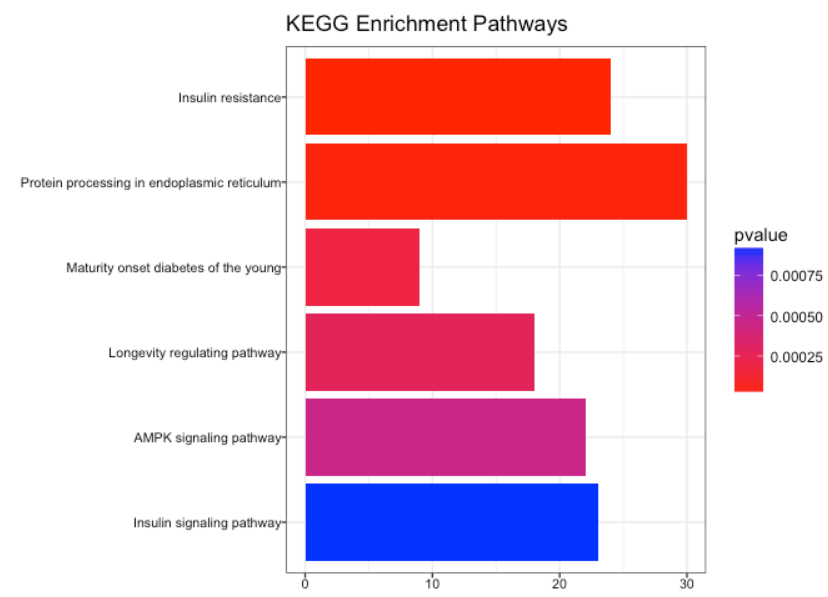
10.1. KEGG
-
使用 KEGG数据库丰富基因
kegg_enrich <- enrichKEGG(gene = names(gene_matrix),
organism = 'mouse',
pvalueCutoff = 0.05,
qvalueCutoff = 0.10)
# 结果可视化
barplot(kegg_enrich,
drop = TRUE,
showCategory = 10,
title = "KEGG Enrichment Pathways",
font.size = 8)
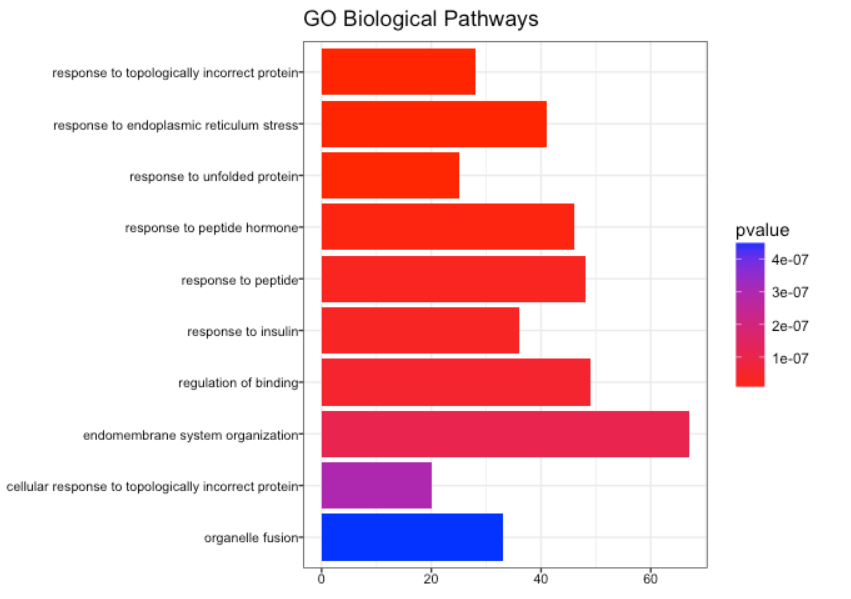
10.2. GO
-
使用 Gene Onotology丰富基因
go_enrich <- enrichGO(gene = names(gene_matrix),
OrgDb = 'org.Mm.eg.db',
readable = T,
ont = "BP",
pvalueCutoff = 0.05,
qvalueCutoff = 0.10)
# 结果可视化
barplot(go_enrich,
drop = TRUE,
showCategory = 10,
title = "GO Biological Pathways",
font.size = 8)
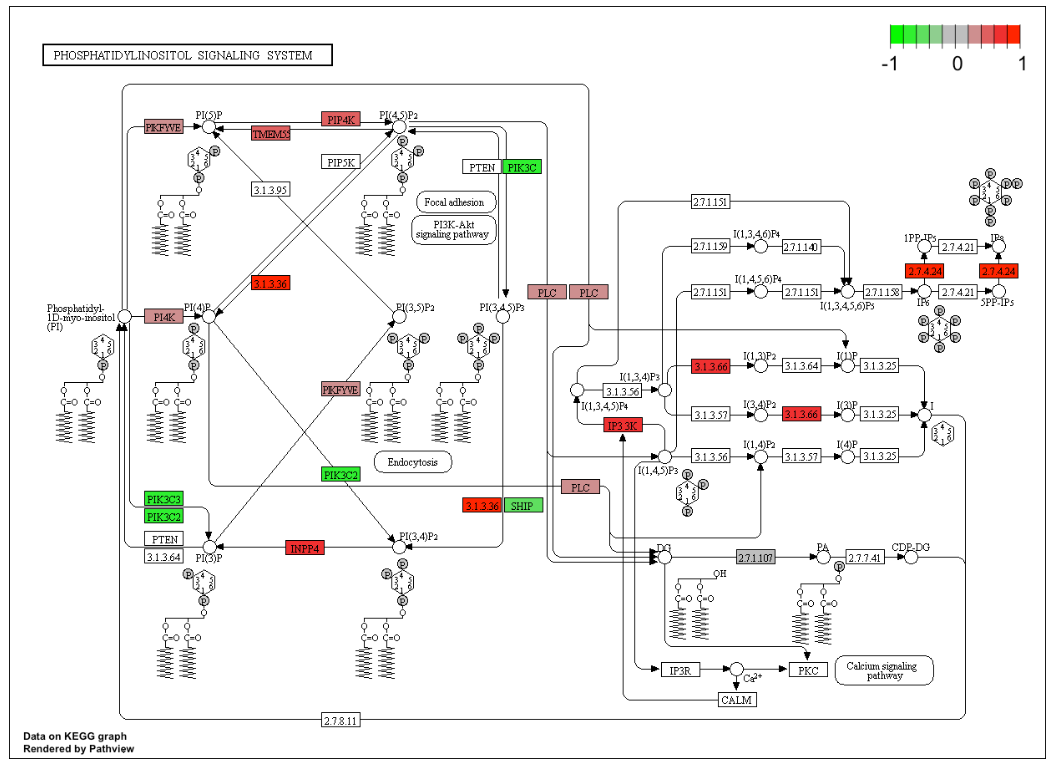
11. 通路可视化
Pathview 是一个包,它可以获取显著差异表达基因的 KEGG 标识符,还可以与 KEGG 数据库中发现的其他生物一起使用,并且可以绘制特定生物的任何 KEGG 途径。
# 加载包
biocLite("pathview") ; library(pathview)
# 可视化通路 (用 fold change)
## pathway.id : KEGG pathway identifier
pathview(gene.data = gene_matrix,
pathway.id = "04070",
species = "mouse")
欢迎Star -> 学习目录
国内链接 -> 学习目录
参考资料
Source: https://github.com/twbattaglia/RNAseq-workflow
本文由 mdnice 多平台发布

![[附源码]计算机毕业设计JAVA基于JSP健身房管理系统](https://img-blog.csdnimg.cn/a7dc37b5521d44f2b51a597c1f69b5e1.png)