-
准备数据:
- 获取基因组序列(FASTA格式)和对应的基因组注释文件(GTF或GFF格式)。
- 获取样本的BAM文件,确保这些文件已经过排序和索引。
- 获取变异信息文件(VCF格式),包含样本的基因型信息。如何获取snp的vcf文件请参考这篇文章:2021.07.30【WGS/GWAS】丨全基因组分析全流程(上)
-
安装GATK工具:
- 下载并安装GATK工具包。确保Java环境已配置好。
-
创建参考序列的索引:
- 使用GATK工具创建参考序列的索引文件。命令如下:
gatk CreateSequenceDictionary -R reference.fasta
- 创建BAM文件的索引:
- 确保BAM文件已经排序并创建索引。命令如下:
samtools sort sample.bam -o sample_sorted.bam
samtools index sample_sorted.bam
注意:早期samtools版本格式在排序步骤命令可能会发生报错,原因是-o的作用是作为输出文件的前缀而不是输出文件。可参考下列命令
samtools sort sample.bam sample_sorted
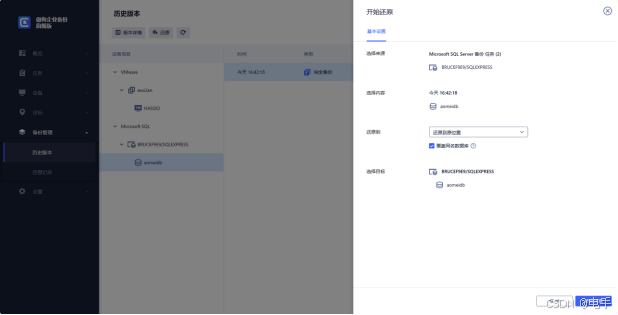
- 运行ASEReadCounter:
- 使用GATK的ASEReadCounter工具进行ASE分析。命令如下:
gatk ASEReadCounter -R reference.fasta -I sample_sorted.bam -V variants.vcf -O output.csv
- 参数说明:
-R:参考基因组序列文件。-I:排序并索引后的BAM文件。-V:变异信息文件(VCF格式)。-O:输出文件,包含ASE分析结果。
- 分析结果:
- 打开输出文件
output.csv,查看每个位点的等位基因特异性读取计数。 - 根据读取计数,计算等位基因的表达水平,进一步分析等位基因特异性表达情况。
- 打开输出文件

通过以上步骤,可以使用GATK ASEReadCounter工具进行ASE分析,详细分析每个位点的等位基因特异性表达情况。
🌟 非常感谢您抽出宝贵的时间阅读我的文章。如果您觉得这篇文章对您有所帮助,或者激发了您对数据科学的兴趣,我诚挚地邀请您:
👍 点赞这篇文章,让更多人看到我们共同的热爱和追求。
🔔 关注我的账号,不错过每一次知识的分享和探索的旅程。
📢 您的每一个点赞和关注都是对我最大的支持和鼓励,也是推动我继续创作优质内容的动力。
📚 我承诺,将持续为您带来深度与广度兼具的数据科学内容,让我们一起在知识的海洋中遨游,发现更多未知的奇迹。
💌 如果您有任何问题或想要进一步交流,欢迎在评论区留言,我会尽快回复您。
🌐 点击下方的微信名片,获取本书资料,加入交流群,与志同道合的朋友们一起探讨、学习和成长。