1.背景
blast作为一种序列相似性比对工具,是生物信息分析最常用的一款软件,必须掌握。不管是做两序列相似性的简单比对,还是引物特异性、序列的来源等个性化分析,都会用到blast比对。许多看似高大上的基因分析,都可归类于序列间的比较,因此blast是生信分析中基础性的工具。
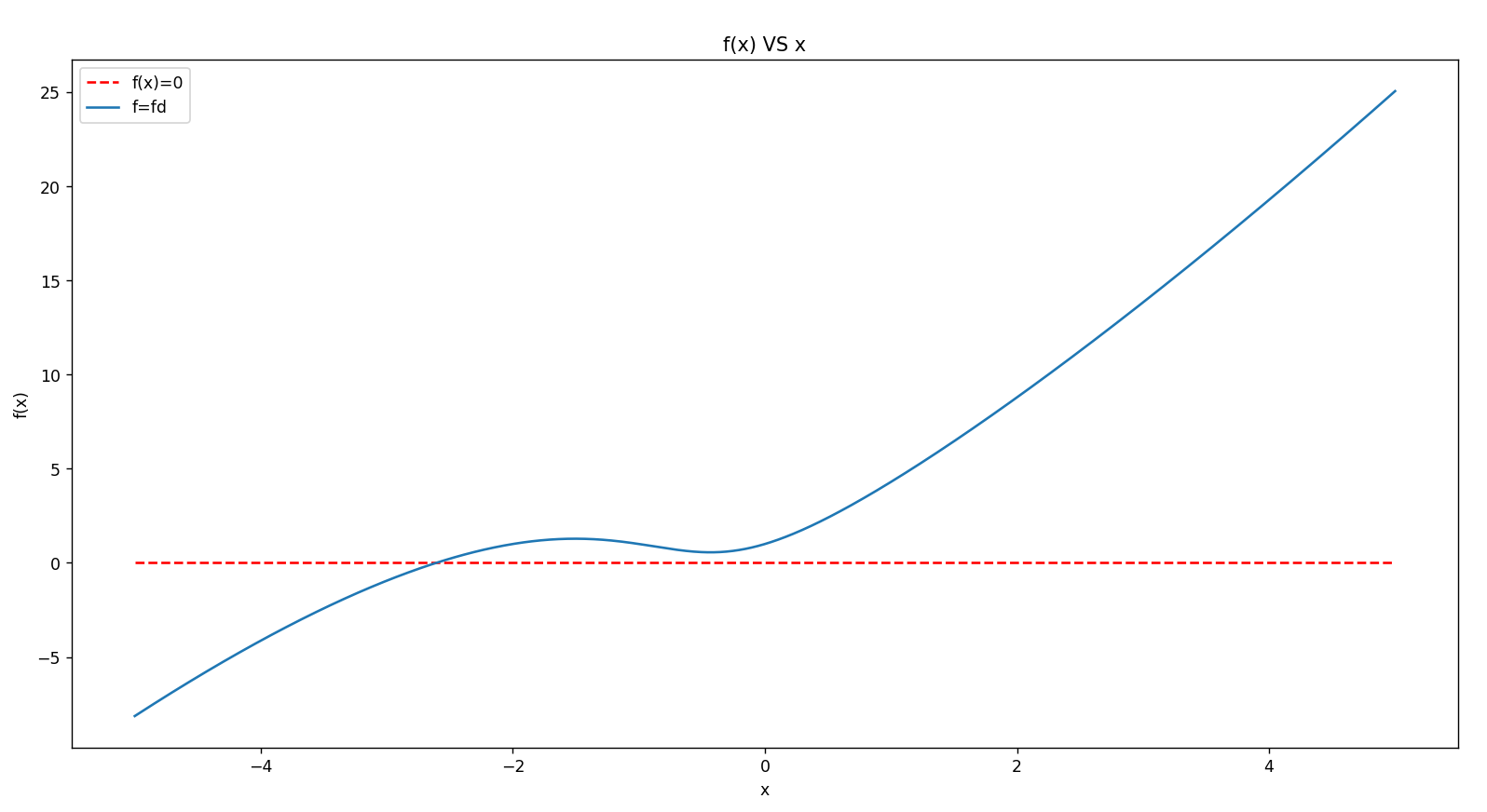
序列比对(Sequence Alignment)的基本问题是比较两个或两个以上序列的相似性。
如果你有一堆测序回来的序列,想要看看它们是来自于哪个物种的,或者想在数据库中搜索对应的同源序列,使用ncbi的在线blast,数据库很全,速度很快,马上就能知道结果。
blastp:蛋白序列与蛋白库作比对,直接比对蛋白序列的同源性。
blastx:核酸序列与蛋白库作比对,将核酸序列先翻译成蛋白序列,再将其与蛋白库作比对。
blastn:核酸序列与核酸库的比对,直接比对核酸序列的同源性。
tblastn:蛋白序列对核算库的比对,现将核酸库翻译成蛋白库,再将蛋白序列与翻译后的蛋白库进行比对。
tblastx:核酸与核酸数据库在蛋白质水平比较
同源性(homology)VS 一致性(identity)
同源性是来描述物种之间的进化关系的,所以在同源性的表达中只能用“有”或者“无”,对于有同源性的物种可以描述为“部分同源”或者“完全同源”。
有些小伙伴们会说“序列A和序列B之间有85%的同源性”,这种说法是不正确的,A和B之间要么有同源性,要么没有同源性,可以这样说:序列A和序列B之间有85%的identity,A和B之间有同源性。
在线blast
blast网站 blsatn_ncbi
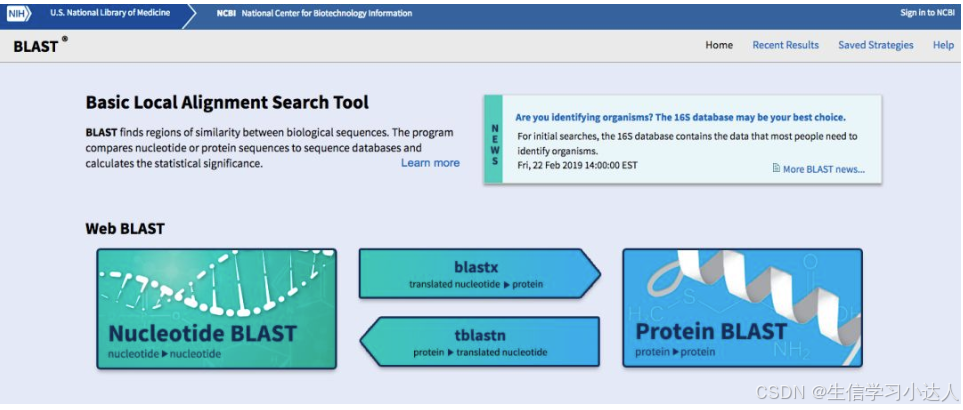
Nucleotide BLAST :核酸序列到核酸库中的一种查询,库中存在的每条已知序列都将同所查序列作一对一的核酸序列比对。
Protein BLAST:蛋白序列到蛋白库中的一种查询,库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。
BLASTX:核酸序列到蛋白库中的一种查询,先将核酸序列翻译成蛋白序列,再对翻译成的每一条序列作一对一的蛋白序列比对。
TBLASTN:蛋白序列到核酸库的一种查询,与BLASTX相反,它是将库中的核酸序列翻译成蛋白质序列,再对所查序列作蛋白与蛋白的比对。
我们点击“Nucleotide BLAST”后就到了blastn的初始界面,输入以下序列
GACGCGGCCGTCGAGGGCGCTCAGGTGGACTTCGACACCGCCAGCCGCATCGAGAGCCGCTACTTCACCCAGCTGGTCACCGGCCAGGTCGCCAAGAACATGATCCAGGCGTTCTTCTTCGACCTCCAGCACATCAACGGCGGCGGCTCCCGCCCCGAGGGCATCGAGCCGGTCAAGATCAACAAGATCGGTGTGCTCGGCGCGGGCATGATGGGCGCCGGCATCGCCTACGTCTCGGCCAAGGC
数据库可以有很多选择,默认是nr/nt库
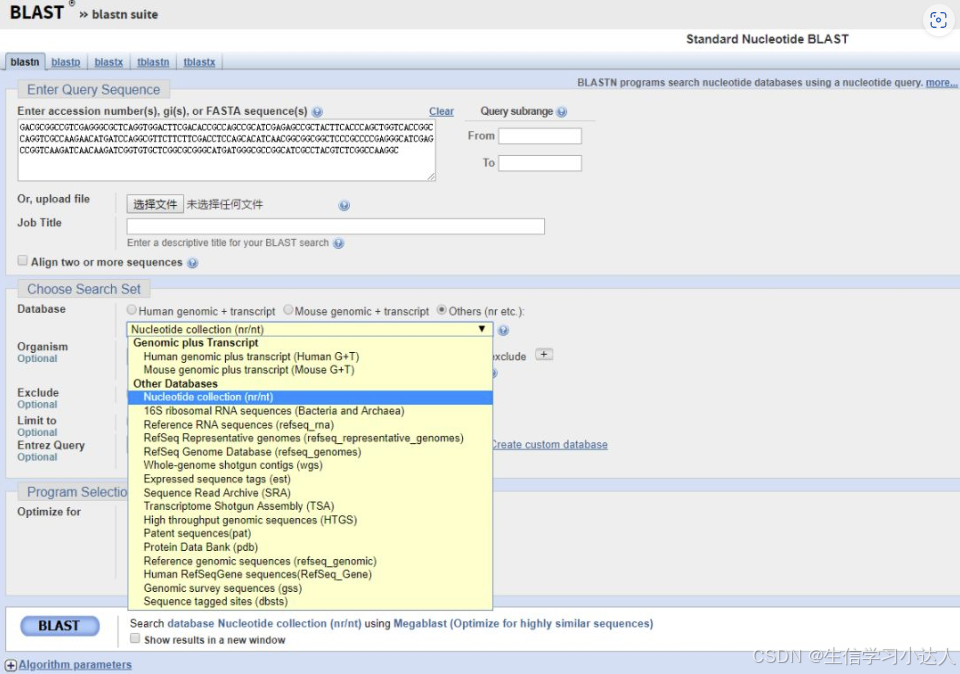
点击“BLAST”即可进行比对,20秒之内就会出现如下结果:
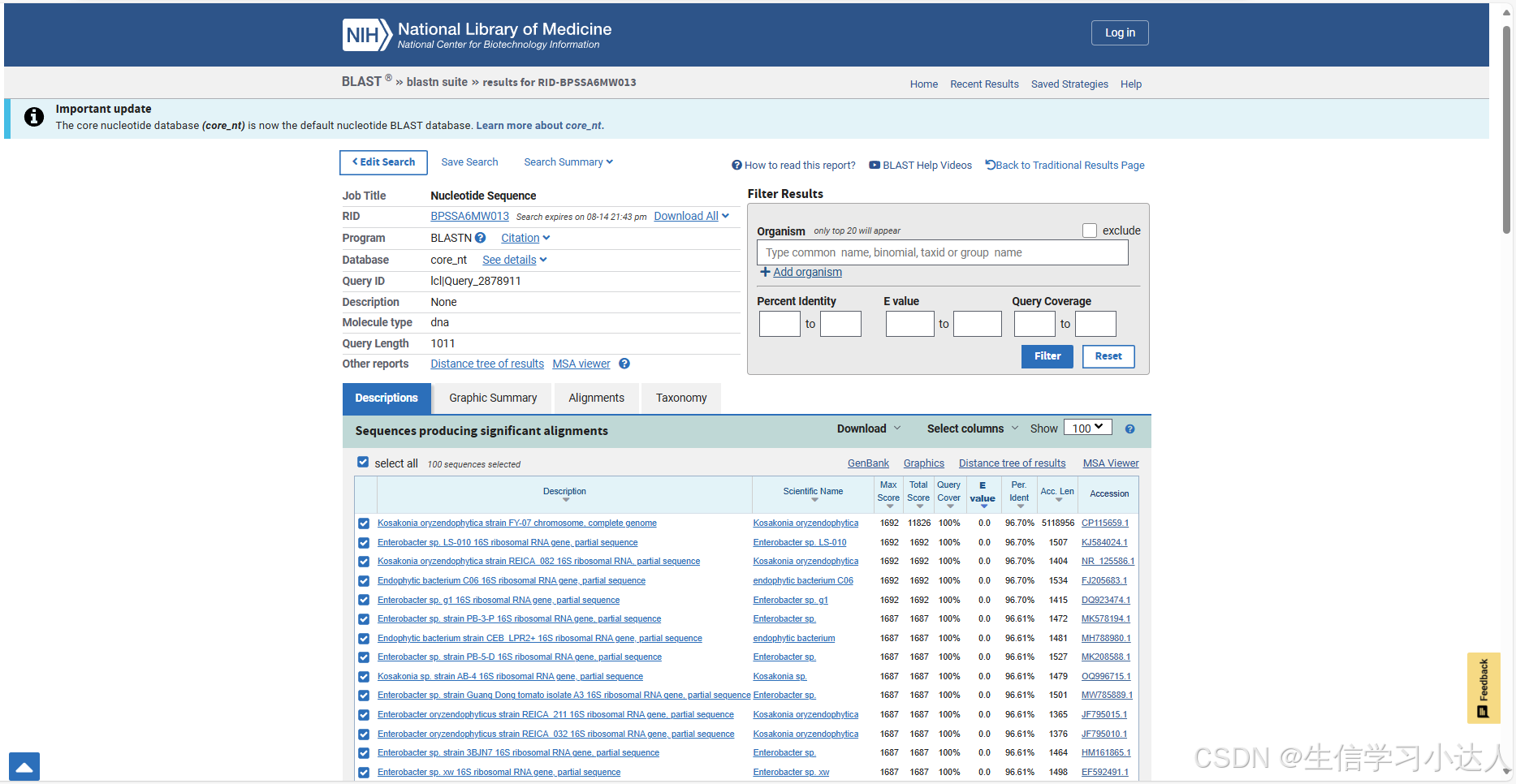
然后点击Expect(E值)和Identities(一致性)值靠前的几个进行比较,筛选出此序列是属于哪个菌的序列

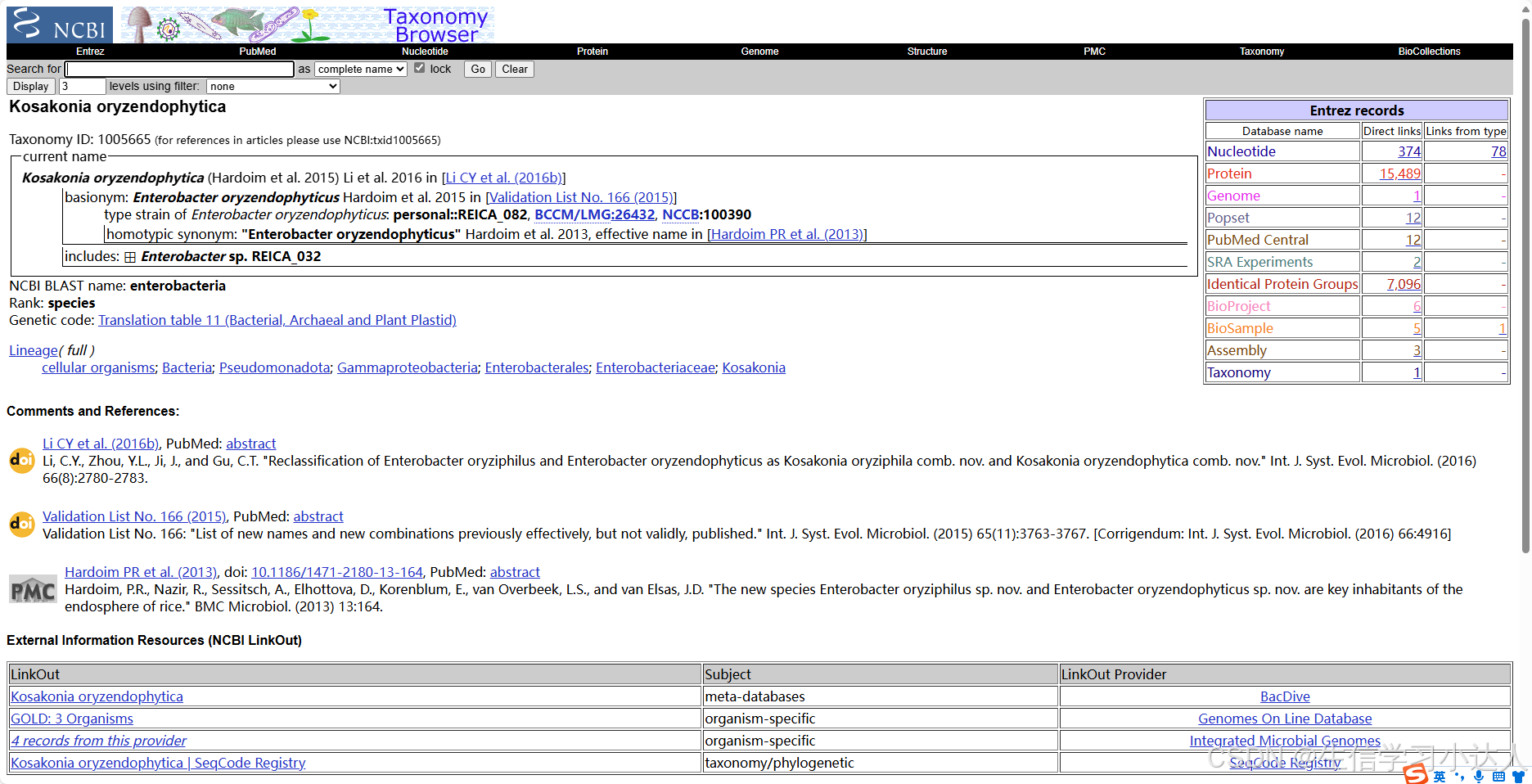
发现这是一段来 Kosakonia oryzendophytica strain FY-07 的序列。
其中,Expect(E值)和Identities(一致性)是评价blast结果的标准。E值接近零或者为零时,说明比上的序列很接近;一致性:匹配上的碱基数占总序列长的百分数。
如果比对其他序列出现如下界面,说明没比上,可以试着选择其他参数
可以选择 Somewhat similar sequences (blastn) 进行再次比对

MEGABLAST : 采用贪婪式算法,多用于比较相似性比较高(相似性在>95%)的序列,灵敏度高,速度快。
Discontiguous MEGABLAST : 灵敏度更高,用于更精确的序列的比对。主要用于跨物种之间的同源比对。
BlastN : 用于比对相似性较差的序列,,相似度较低的序列也可以查找到,比对结果最多,速度最慢。 但允许更短序列的比对(如短到7个碱基的序列),例如做短的引物的比较可以使用这个选项。
如果想比较手头上的序列A和序列B之间的相似性,可以在blastn的初始界面点击
Align two or more sequences,就可以分别放入序列进行比对了。

参考文献来源:
生信入门:序列比对之blast在线和本地使用_生物信息学blast-CSDN博客