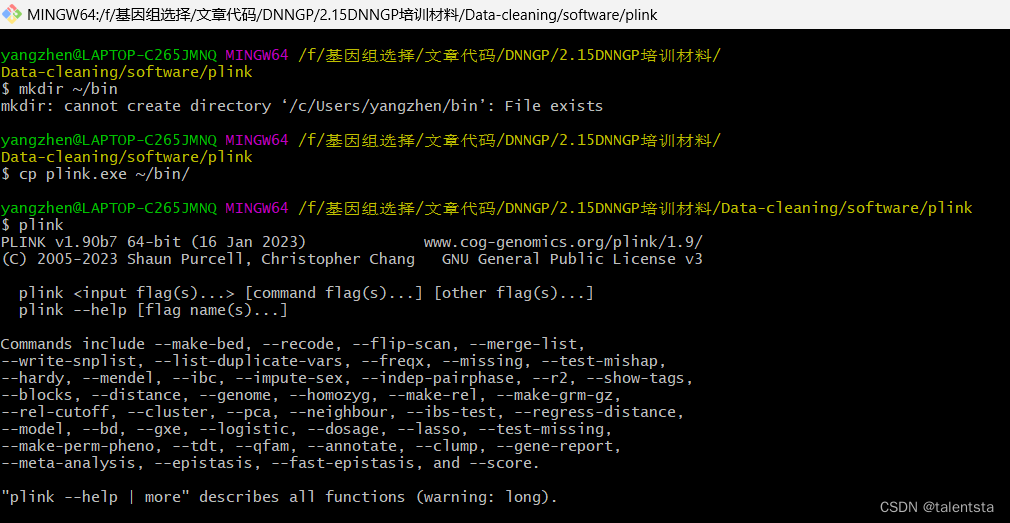
首先使用git进行plink环境配置,显示环境安装成功,在此环境下可以使用plink
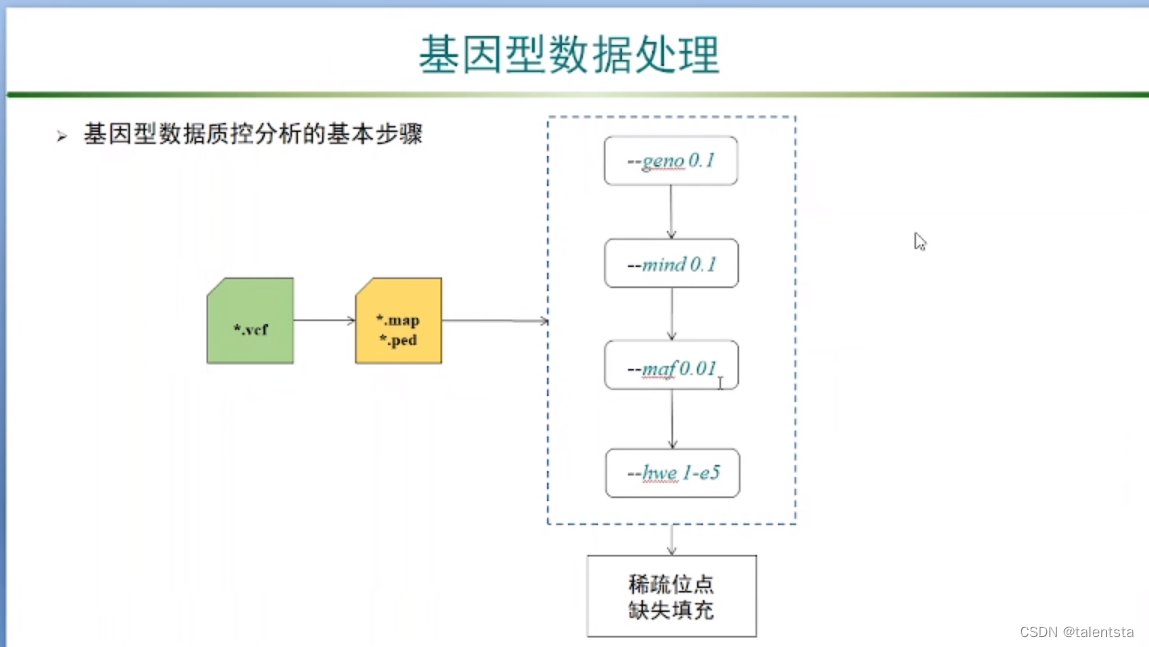
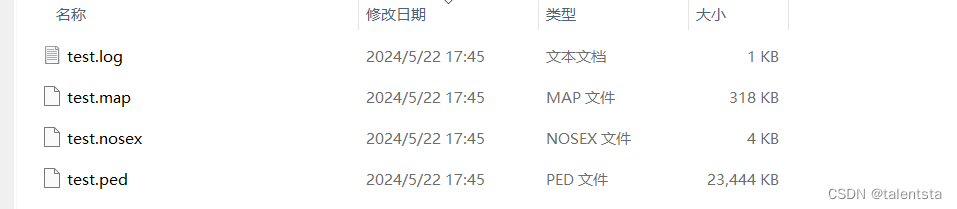
在基因型数据处理过程中,看到vcf文件后首要做的就是将vcf文件转成二进制文件,输入命令 plink --vcf genotype.vcf --allow-extra-chr --recode --out test 之后,转化为了如下图所示四种类型的文件,其中有map和ped文件。
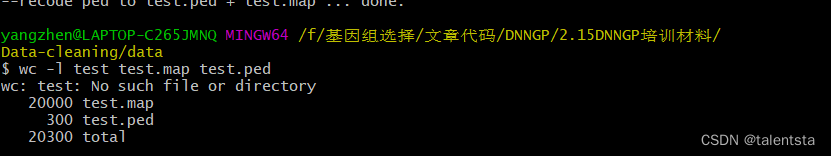
 通过 wc -l test test.map test.ped 查看样本的数量,这里是有20000个SNP和300个样本,we -l test 是查看基本情况,后边接想要查看的文件
通过 wc -l test test.map test.ped 查看样本的数量,这里是有20000个SNP和300个样本,we -l test 是查看基本情况,后边接想要查看的文件
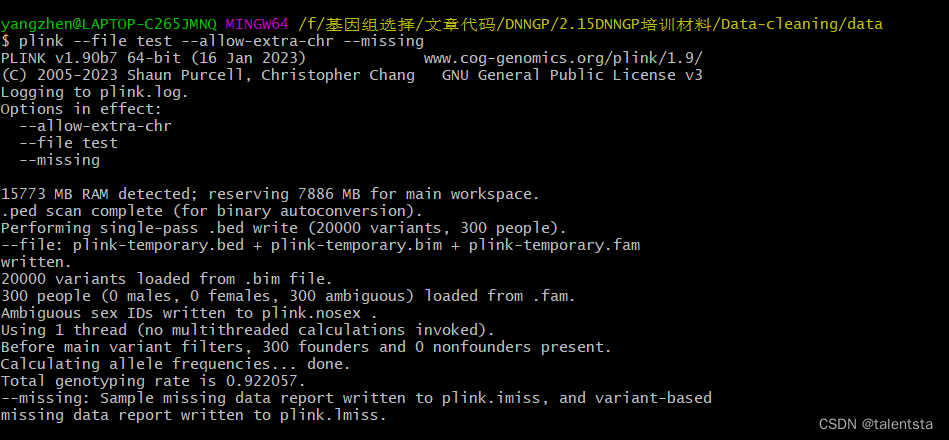
通过执行命令 plink --file test --allow-extra-chr --missing 检查基因型和样本缺失值情况,命令显示缺失值情况文件已被写入plink.lmiss文件
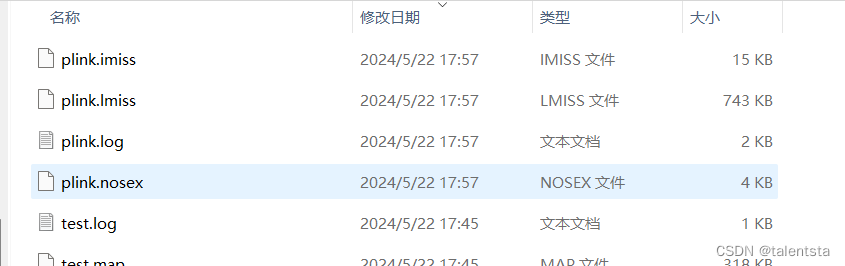
根据提示,可以查看缺失的数据有哪些,通过执行命令 head plink.imiss 和 head plink.lmiss ,也可以查看缺失率

setwd("F:/基因组选择/文章代码/DNNGP/2.15DNNGP培训材料/Data-cleaning/data")#设置工作路径
library(data.table)
library(openxlsx)
library(sommer)
library(dplyr)
library(ggplot2)
library(VIM)
library(mice)
library(zoo)#标记缺失
snpmiss<-read.table(file="plink.lmiss",header = TRUE)
dim(snpmiss)
head(snpmiss)
hist(snpmiss[,5],main="Histogram SNP missingness",xlab = 'SNP miss rate',
ylab='Frequency',col = '#E47D28',cex=0.5,pch=19,
font.lab = 2,cex.lab = 1.2,ylim=c(0,4000))
#个体缺失
indmiss<-read.table(file="plink.imiss",header = TRUE)
dim(indmiss)
head(indmiss)
hist(indmiss[,6],main = "Histogram individual missingness",
xlab = 'Individual miss rate', ylab='Frequency',
col = '#753281',cex=0.5,pch=19,
font.lab = 2,cex.lab = 1.2,ylim=c(0,150))通过执行R代码,可以对查看的标记和个体缺失进行可视化分析。
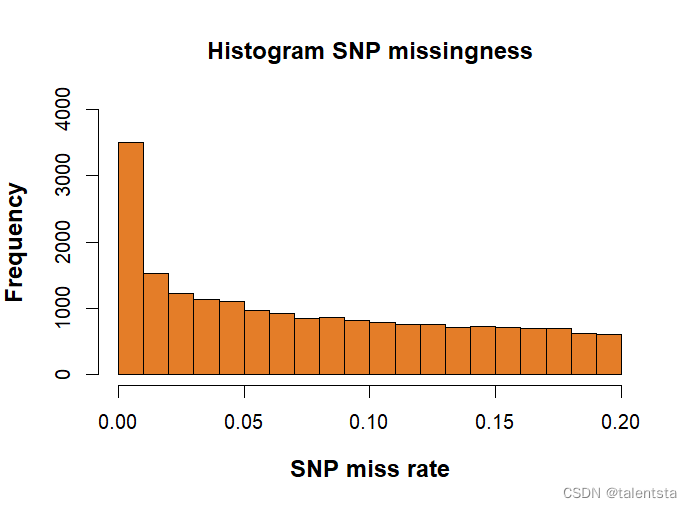
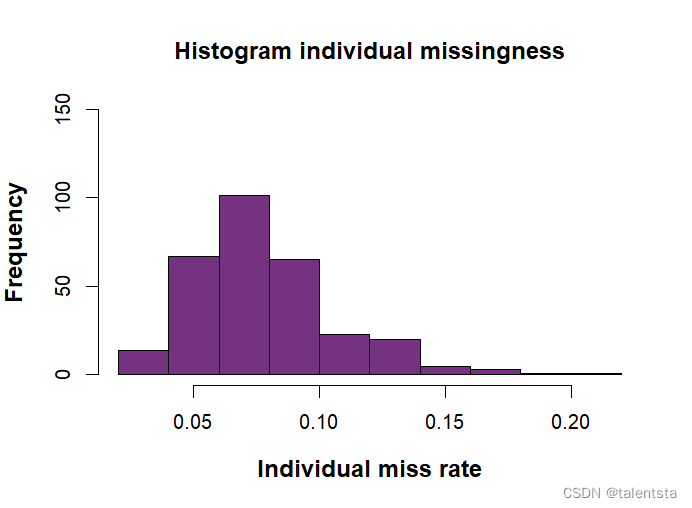
检查完基因型和样本缺失值情况后,执行命令
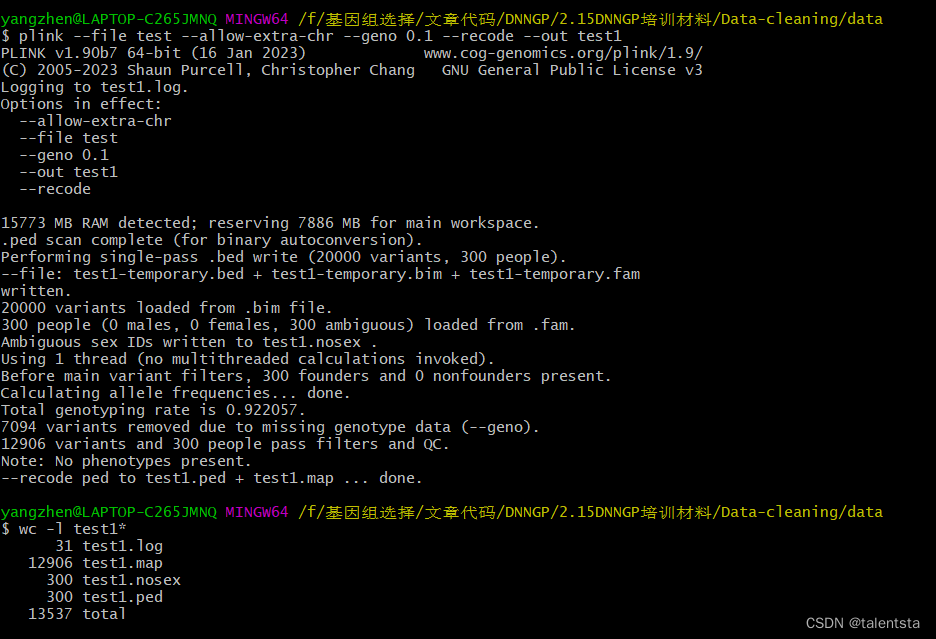
plink --file test --allow-extra-chr --geno 0.1 --recode --out test1
对基因型进行过滤输出为test1,同时对过滤后的文件查看其SNP样本数量变为了12906
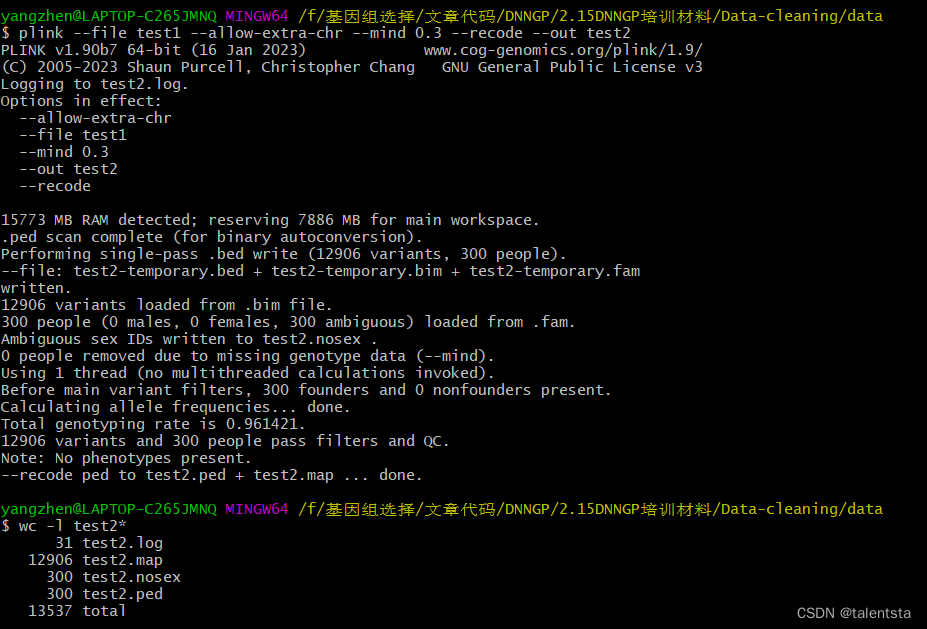
 对个体执行缺失控制,执行命令
对个体执行缺失控制,执行命令
plink --file test1 --allow-extra-chr --mind 0.3 --recode --out test2
输出为test2,这里设置的缺失数较大,故没有对其删除,还是和刚才的样本量相同