文献介绍

文献题目: Spatial multi-omics at subcellular resolution via high-throughput in situ pairwise sequencing
研究团队: 曹罡(深圳理工大学)、戴金霞(华中农业大学)
发表时间: 2024-05-14
发表期刊: Nature Biomedical Engineering
影响因子: 28.1
DOI: 10.1038/s41551-024-01205-7
摘要
空间多组学技术有助于发现细胞功能和疾病机制的新见解。在这里,我们报告了多组学原位成对测序 (MiP-seq) 的开发和应用,这是一种以亚细胞分辨率同时检测 DNAs、RNAs、蛋白质和生物分子的方法。与其他原位测序方法相比,MiP-seq 增强了解码能力,降低了测序和成像成本,同时保持了检测基因突变、等位基因特异性表达和 RNA 修饰的效率。MiP-seq 可以整合体内钙成像和拉曼成像,这使我们能够生成小鼠脑组织的空间多组学图谱,并将基因表达与神经元活动和细胞生化指纹相关联。我们还报告了一种连续稀释策略,用于在原位测序过程中解决光学拥挤信号。高通量原位成对测序可以促进组织分子和功能图谱的多维分析。
前言
多细胞生物优雅地协调特定细胞中适当基因的共转录。这些空间转录谱控制不同细胞的功能,以完成复杂的生理任务。全面空间组学的描述将为理解特定细胞的分子功能铺平道路,并通过例如在癌症治疗前表征免疫微环境来极大地推进精准诊断。
最近,最先进的空间转录组学方法已被开发用于高度多重 RNA 原位检测。一般来说,这些空间转录组学方法是基于三种策略开发的:多重荧光原位杂交(FISH)(如 SeqFISH 和 MERFISH)、原位测序(如 ISS、FISSEQ、STARmap、INSTA-seq 和 ExSeq) 、原位捕获与高通量测序相结合(如 Slide-seq、HDST 和 DBiT-seq)。然而,空间组学技术仍处于起步阶段。例如,大多数方法只能破译一种生物分子的空间信息,多种生物分子的原位联合检测仍然具有挑战性。此外,由于大多数基于原位测序和基于 FISH 的方法仅具有 (4 代表四个荧光基团,N 代表测序或杂交轮数)解码能力,因此需要多轮测序才能实现高通量空间组学。需要提高捕获效率、通量、细胞分辨率和光学拥挤以及降低这些实验成本的方法。在这里,我们报告了一种高通量靶向原位测序方法,多组学原位成对测序(multi-omics in situ pairwise sequencing, MiP-seq),的开发,能够以亚细胞分辨率有效检测脑组织中的多重 DNAs、RNAs、蛋白质和生物分子。
研究结果
1. MiP-seq 在生物分子空间分析中的应用
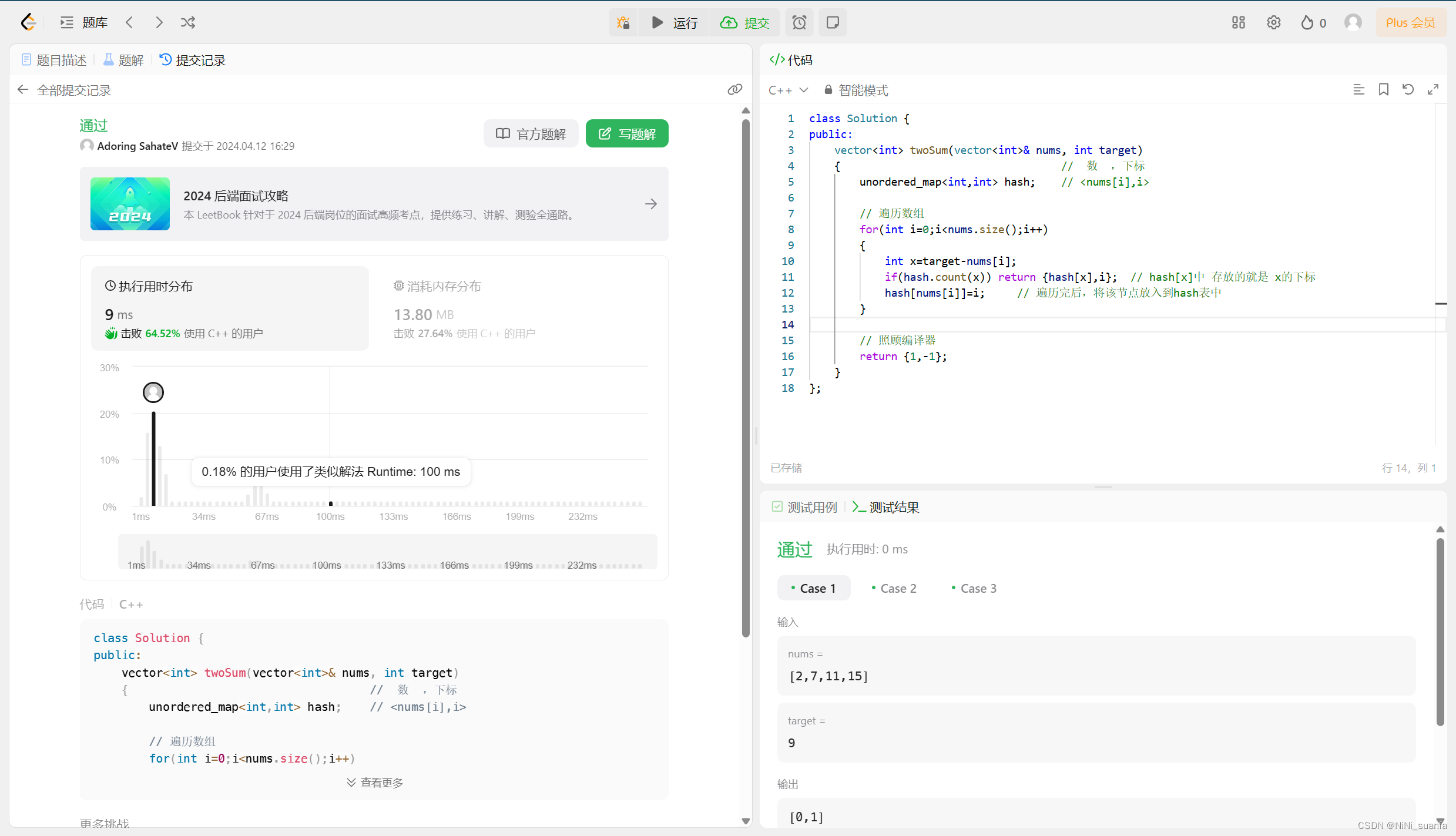
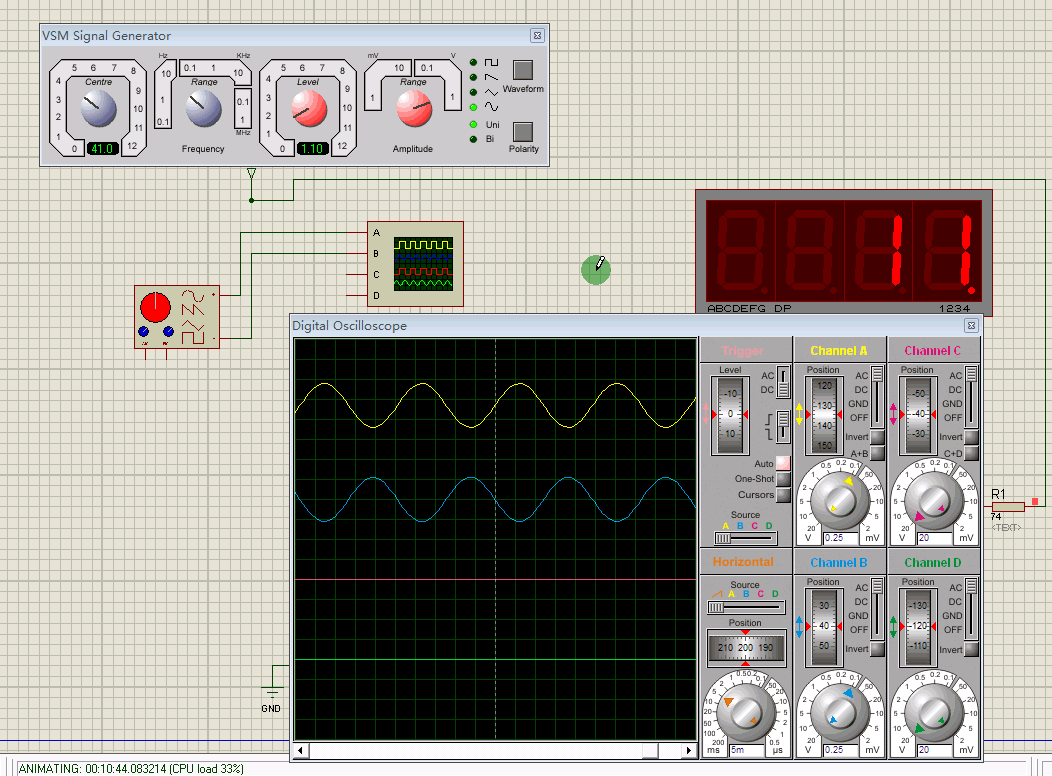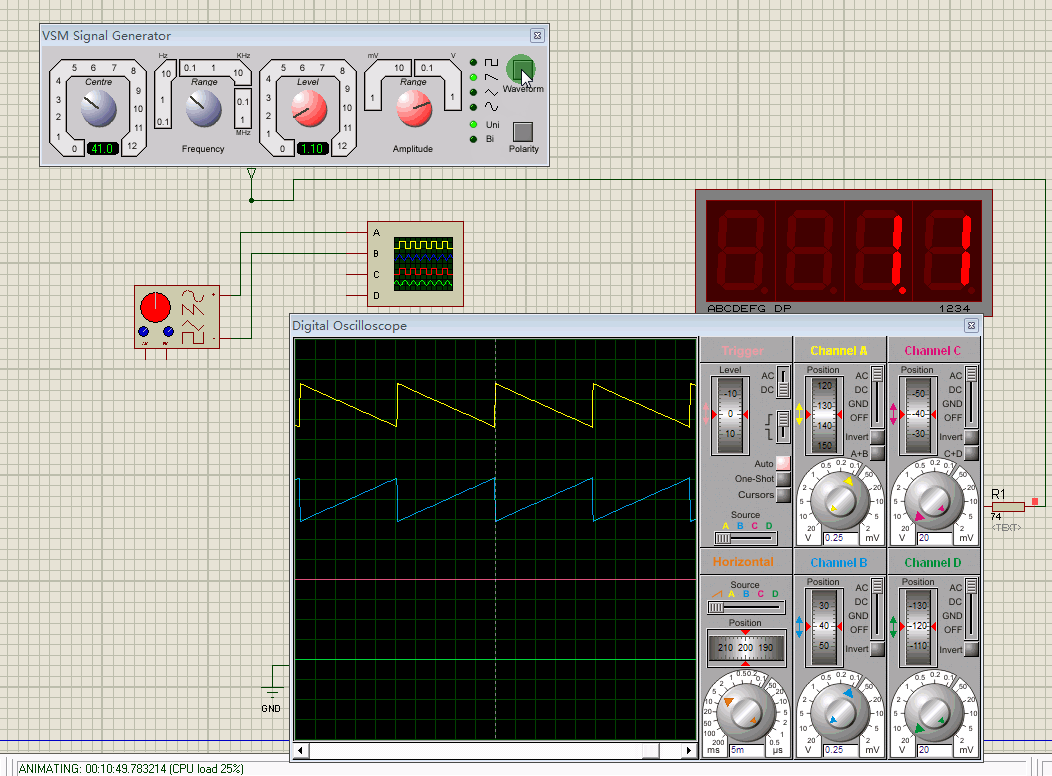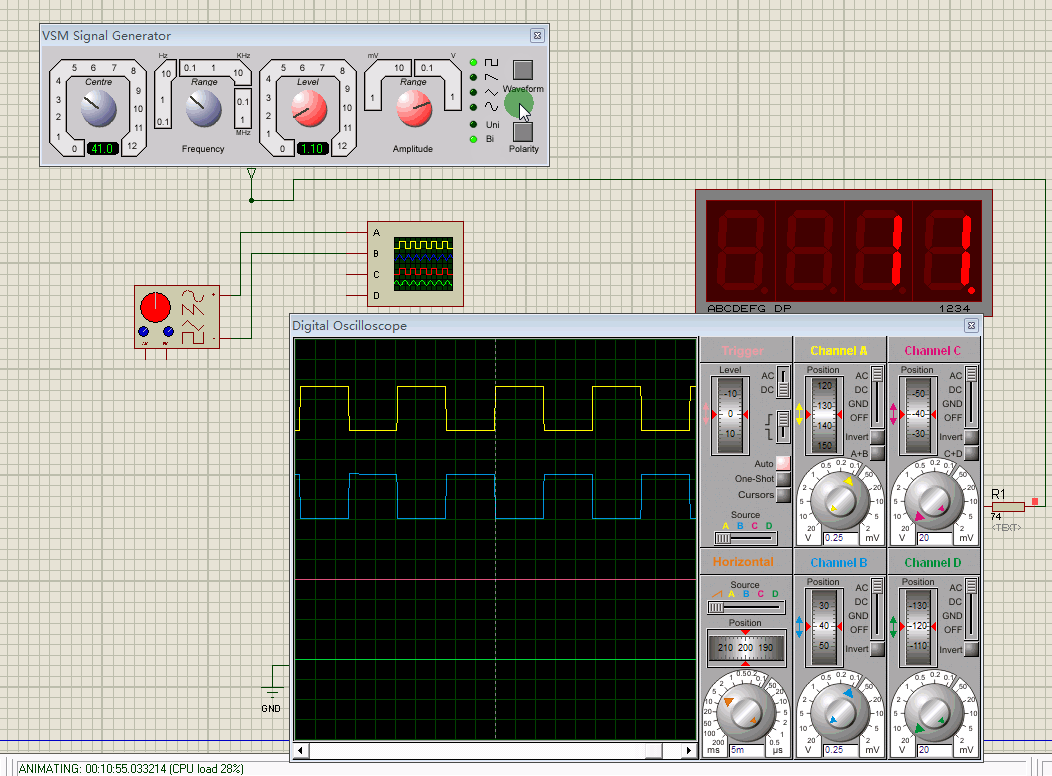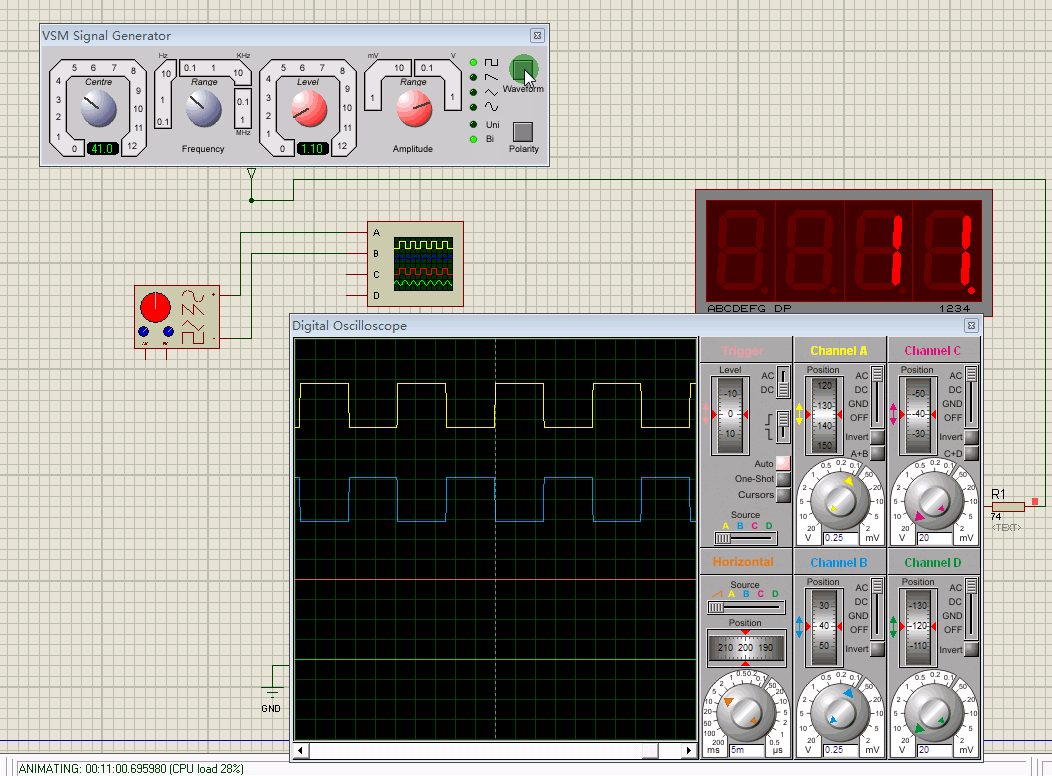
MiP-seq 对 DNA 和 RNA 的原位检测是通过针对核酸的直接锁式探针实现的,而蛋白质和生物分子的检测是通过检测抗体偶联核酸的锁式探针实现的(Fig. 1a)。首先,我们将 MiP-seq 应用于空间 RNA 分析,该过程涉及锁式探针杂交、原位滚环扩增 (RCA) 和基于连接的双条形码测序(Fig. 1b)。值得注意的是,每个锁式探针包含两个条形码(B1 和 B2),这增加了其信号解码能力。根据之前的研究,我们比较了不同的连接酶(Supplementary Fig. 1)并选择 SplintR 来连接 RCA 模板,这提高了 RCA 模板连接效率。这种连接方法依赖于精确匹配的碱基配对,从而可以通过 MiP-seq 检测目标 RNA 中的单核苷酸多态性 (SNP)。由于锁式探针和起始引物(IP)的目标依赖性连接与目标 RNA 和锁式探针互补,RCA 与目标一起牢固地在原位进行(Fig. 1b)。为了进行高通量检测,通过连接和信号解码对原位滚环扩增产物(RCPs)进行多轮测序(Fig. 1b)。
a. MiP-seq 在空间组学分析中的应用图。
b. 用于 RNA 原位检测的 MiP-seq 流程图。I. 双条形码引物 (BP) 和起始引物 (IP) 与目标 RNA 杂交。II. BP 和 IP 与目标 mRNA 退火形成锁式结构。III. BP 通过 RNA 依赖性 DNA 连接酶 SplintR 环化形成 RCA 模板。IV. 圆形 BP 的 RCA 形成滚环产物。V. 双锚定引物在双条形码序列旁边杂交。随后,通过连接测序,荧光标记的查询探针对条形码序列进行查询。VI. 每轮测序解码 10 个双条形码读数,N 轮后可查询
个编码。
c. 根据每轮测序的配对碱基的合并荧光信号进行十个双条形码查询的示意图。细虚线表示配对碱基 AA。粗虚线表示配对的碱基 AG 和 GG。虚线表示配对的碱基 AC、GC 和 CC。
d. 十个双条形码每一个的合并荧光信号。
e. 双条形码序列查询示例。方框中的点显示了三轮中每一轮相应的条形码读数。
f. 底部:C57BL/6 雌性小鼠与 BALB/c 雄性小鼠杂交后代海马中 Fam171b、Hba-a2、Rpl15 和 Rps11 等位基因特异性基因表达的原位测序。上图:海马中每个细胞(n = 6,084 个细胞)的 Fam171b、Rps11、Hba-a2 和 Rpl15 等位基因的表达水平(每个细胞的 RCPs)。RH,右侧海马体。用 oligo-dT-Cy3(灰色)染色的细胞形态与相应的基因表达谱重叠。等位基因特异性基因表达的检测重复至少三次。
g. m6A 甲基化 RNA 和非甲基化 RNA 转染 HEK-293T 细胞,可通过 MiP-seq 进行区分。使用 Student's t-test (two-tailed) 对来自 25 个成像视野的非甲基化和甲基化 RNA 进行统计分析(数据表示为 mean ± s.d., ****P < 0.0001;原始数据可在 Supplementary Table 3 中找到)。显示了 RCPs(Cy3 红色荧光)和 DAPI(蓝色荧光)。m6A 甲基化 RNA 和非甲基化 RNA 的检测重复至少 3 次。Scale bars, 2 µm in e, 50 µm in f, 15 µm in g。
为了提高信号解码通量,我们在每一轮中同时对双条形码碱基进行测序。在该策略中,四个荧光信号的两两组合可以在每个循环内对十种不同的组合进行编码(Fig. 1b–d)。因此,可以通过 N 轮测序解码 个基因(Fig. 1b),与其他原位测序方法(通常可以解码 个基因)相比,这在显着减少的测序轮次中显着提高了基因检测通量。由于 RCA,可以在双条形码测序的基础上清楚地识别和解释以点表示的 RCP 信号(Fig. 1d,e)。为了进行双条形码测序,我们尝试了两种策略,双端测序和成对测序(Supplementary Fig. 2a,b)。在双端测序策略中,通过单锚定双末端连接测序对双条形码碱基进行测序(Supplementary Fig. 2a)。分析共现和相关性,以验证人脑微血管内皮细胞(HBMEC)中重叠双条形码的百分比(Supplementary Figs. 2c–e, 3a–c, 4a–c and 5a–c)。发现双条形码荧光信号的强度高度相关,表明双条形码测序方法的可靠性(Supplementary Figs. 2d, 3b, 4b and 5b)。这些数据通过双荧光信号的共现分析进行了验证(Supplementary Figs. 2e, 3c, 4c and 5c)。
为了获得最大的双碱基识别准确性,我们进行了成对测序来破译双条形码,并将该方法的效率与双端测序策略的效率进行了比较(Supplementary Fig. 2a,b)。如 Supplementary Figs. 2f–h, 3d–f, 4d–f and 6a–c 中所示,成对测序比双端测序策略产生更大的重叠。这些数据表明,我们基于成对测序的 MiP-seq 方法除了高编码能力之外,还可以实现更准确的双碱基识别。随着高灵敏度显微镜技术和深度学习辅助成像分析的快速发展,我们相信双碱基识别的准确性可以进一步提高。
接下来,我们通过比较 MiP-seq 与基于第三代杂交链式反应(HCR3.0-FISH)的 FISH 对人类 GAPDH 和 MTOR 基因转录本的检测程度来测试 MiP-seq 在 RNA 检测方面的效率 (Supplementary Fig. 7)。首先,对于 MiP-seq,使用了针对 GAPDH 和 MTOR 转录本的 2 个目标探针,每个细胞的平均检测效率达到 HCR3.0-FISH 的约 40%。通过将 MiP-seq 靶向探针的数量增加到 8 个探针,GAPDH 和 MTOR 基因转录本检测的效率分别达到 HCR3.0-FISH 的 96% 和 56%。与非靶向原位测序方法(例如 FISSEQ)相比,MiP-seq 在 RNA 检测中表现出更高的效率,每个细胞中低表达基因(包括 YWHAZ、FUS、HPTR1 和 B2M)的 spot 计数更多(Supplementary Fig. 8)。此外,MiP-seq 检测到的小鼠大脑中不同基因的表达模式与根据 Allen 研究所脑图(Extended Data Fig. 1a,b)通过传统原位杂交揭示的模式相似。此外,MiP-seq 能够以亚细胞分辨率检测基因转录本,如细胞质中的 Kif5a mRNA 信号和细胞核中的 Snhg11 mRNA 信号的结果所示(Extended Data Fig. 1c)。
为了进一步证实 MiP-seq 在多重 RNA 检测中的保真度,我们比较了 MiP-seq 和 MERFISH 检测到的基因表达模式。为此,我们下载了已发表的 MERFISH 数据,并从 MERFISH 基因列表中随机选择 77 个基因用于同一大脑区域(小鼠下丘脑视前区)的 MiP-seq。比较分析揭示了 MiP-seq 和 MERFISH 之间可比较的基因表达趋势(Extended Data Fig. 2),表明 MiP-seq 在多重 RNA 检测中具有高保真度。
MiP-seq 严格依赖于与目标序列精确匹配的碱基对的相邻连接来形成 RCA 模板。因此,它可以进一步用于检测具有单碱基变异和携带 RNA 修饰的靶标。如 Extended Data Fig. 3a 所示,我们进行了 MiP-seq 以区分共培养的 HBMEC 细胞(人类来源)和 Bend3 细胞(小鼠来源)中具有单核苷酸变异的人类和小鼠 β-actin mRNA。此外,我们利用 MiP-seq 分析小鼠大脑中的多重等位基因特异性基因表达模式。首先通过 RNA-seq 鉴定亲本基因(Fam171b、Rps11、Hba-a2、Rpl15)的 SNPs,然后将 MiP-seq 应用于小鼠海马和下丘脑室旁核(PVN)的样本。MiP-seq 一致检测到亲本基因的不均匀等位基因特异性基因表达模式;例如,Rps11 基因的 G 等位基因以及 Rpl15 和 Hba-a2 基因的 T 等位基因,它们优先在海马和 PVN 中表达(Fig. 1f and Extended Data Fig. 3b–d)。这些数据可能意味着同一细胞中的母本和父本染色体可能正在经历不同的基因转录调控过程。
由于 RNA 修饰可能会破坏 RCA 模板连接,因此我们假设 MiP-seq 可用于区分甲基化 RNA 和未修饰 RNA。为了验证这一假设,将合成的 N6-甲基腺苷 (m6A) 修饰的 RNA 和未经修饰的对照 RNA 转染至 HEK-293T 细胞中并进行 MiP-seq。如 Fig. 1g 所示,我们观察到未修饰 RNA 的阳性信号,而由于甲基化核苷酸位点的相邻连接失败,我们检测到 m6A 修饰 RNA 的可忽略不计的信号。因此,MiP-seq 与其他表观遗传修饰测序信息相结合,有可能用于区分特定位点的 m6A 甲基化 RNA。此外,我们使用 MiP-seq 同时检测肿瘤中多个基因的野生型(WT)和点突变。根据我们的 RNA-seq 数据,我们将 MiP-seq 应用于 WT,并在患有 C6 神经胶质瘤的大鼠大脑中突变 Eno1、Nptxr、Spp1、Tnc、Tuba1a 基因(Extended Data Fig. 3e)。在一轮原位测序中通过 10 个条形码读数同时检测到这些神经胶质瘤组织中突变体和 WT 基因的表达(Extended Data Fig. 3e)。
2. 多个基因的大视野 3D 重建和整体空间分析
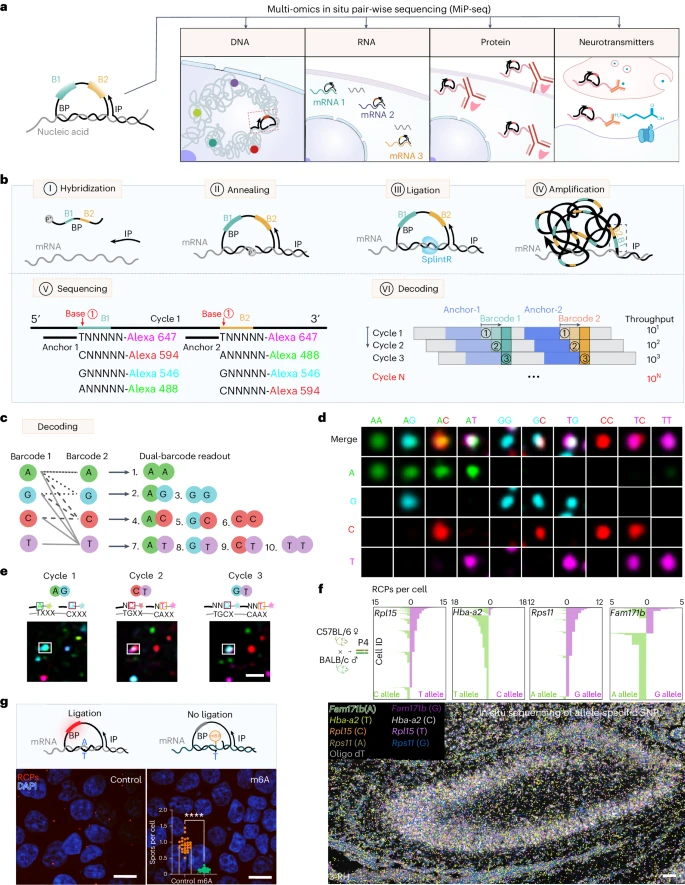
接下来,我们尝试利用 MiP-seq 实现大成像视野的多基因检测。如 Fig. 2a 所示,通过一轮 MiP-seq 描绘了小鼠大脑整个矢状切片中 10 个基因(Calb1、Gad1、Plp1、Neurod1、Cck、Lamp5、Ndnf、Pax2、Pcp4 、Penk)的空间转录组图谱。重要的是,MiP-seq 检测到的这些基因的表达模式与 Allen 脑科学研究所的结果一致 (Fig. 2b and Extended Data Fig. 4a,b)。为了进一步构建三维(3D)基因表达架构,通过 MiP-seq 对整个小脑间隔 200μm 的 34 个矢状切片进行多重基因检测(Fig. 2c,d and Supplementary Video)。重建图像显示了整个小脑中 Calb1、Gad1、Neurod1 和 Plp1 基因表达和分布的 3D 视图(Fig. 2e and Extended Data Fig. 4c)。此外,我们还优化了 MiP-seq,用于全组织中的多重基因检测。如 Fig. 2f,g 所示,在整个斑马鱼端脑中检测到 foxg1a、hoxb3 和 gad1b 基因的原位基因表达模式。
a. 小鼠大脑整个矢状切片中十个基因(Calb1、Gad1、Plp1、Neurod1、Cck、Lamp5、Ndnf、Pax2、Pcp4、Penk)的空间转录组图谱。小脑和海马体中的虚线矩形分别被放大在 b 和 Extended Data Fig. 4b 中查看。
b. 左:a 中矩形区域的小脑十个基因共同检测放大图。中和右:MiP-seq 检测到的小脑基因表达模式与 Allen 脑科学研究所的结果一致。
c-e. 整个小脑中多重基因(Calb1、Gad1、Neurod1、Plp1)检测的荧光图像 3D 重建流程图。从整个小脑中选取间隔 200μm 的 34 个矢状切片,通过 MiP-seq 和成像进行多重基因检测,然后进行重建。
f-g. MiP-seq 检测的整个斑马鱼端脑中 foxg1a、hoxb3、gad1b 基因表达和分布的 3D 视图。a-g 中这些基因的检测至少重复了 3 次。Scale bars, 1,000 µm in a, 300 µm in b, 500 µm in e, 30 µm in f and g。
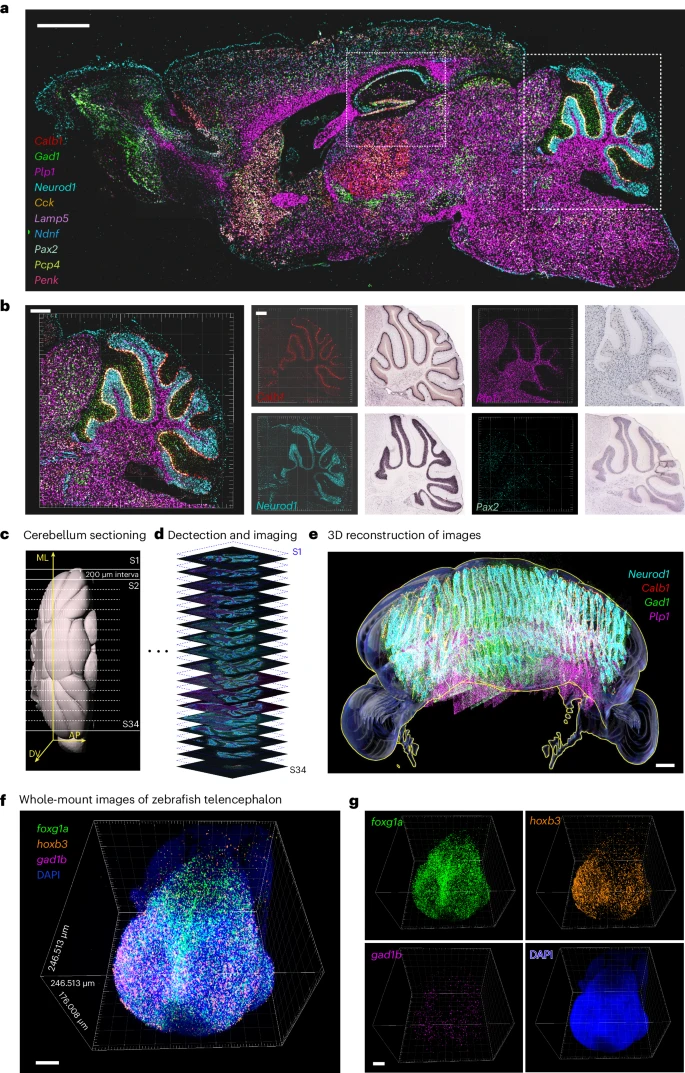
3. 使用 MiP-seq 对 HDB 核中 100 个基因进行空间转录组分析
由于 MiP-seq 的高通量能力,我们通过 2 轮测序绘制了下丘脑对角带水平肢(HDB)核内 100 个基因的空间转录组图谱(Fig. 3a)。高通量图像处理涉及图像配准、候选点识别、解码、细胞分割和基因分配。用于高通量成对双色信号解码的空间转录组分析流程如 Extended Data Fig. 5 所示。我们的数据揭示了 HDB 中 100 个基因的单细胞分辨率空间分析,包括神经递质相关基因、神经元标记基因和神经元活动标记基因(Fig. 3a and Extended Data Fig. 6a,b)。在 100 个基因图谱的基础上,确定了肽激素基因、神经递质相关基因及其受体基因的独特空间表达模式(Fig. 3b and Extended Data Fig. 6c)。通过空间基因共表达分析,我们发现某些基因在空间上共表达(Fig. 3c)。例如,观察到 Snap25 和 Scg2 表达的高度空间相关性(Fig. 3d,e),这与它们在突触小泡胞吐作用和突触形成中的共同功能一致。有趣的是,我们还鉴定了一些具有某些空间互斥表达模式的基因,例如 Hap1 和 Pdgfra(Extended Data Fig. 6d,e)。与此相一致的是,之前的研究表明 Hap1 在神经元中表达富集,Pdgfra 在少突胶质细胞祖细胞中特异性表达。
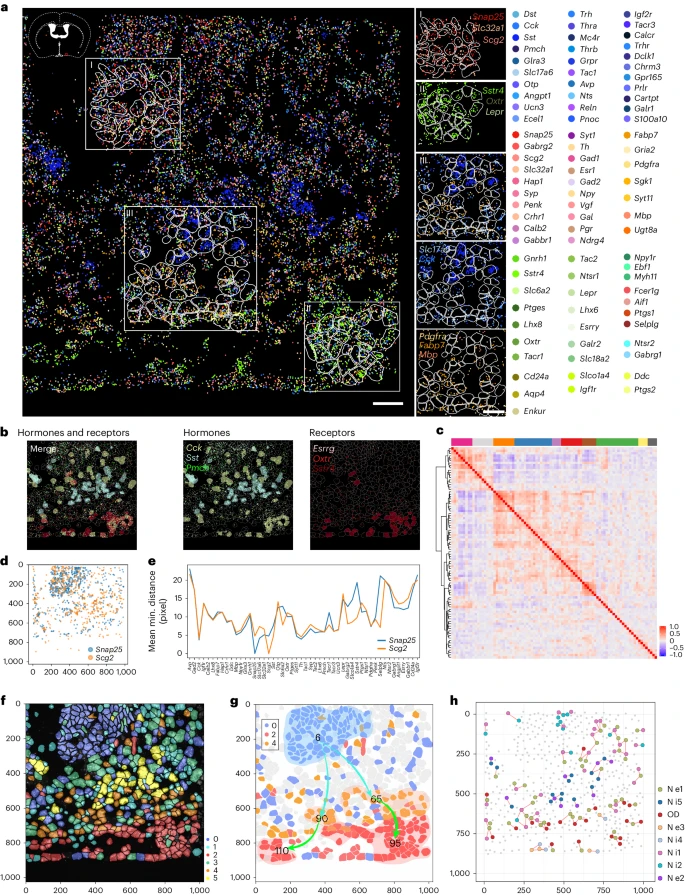
a. 左:MiP-seq 检测到的 HDB 中 100 个基因的空间转录组。每个基因的转录本都是伪彩色的。右图:左侧标有矩形(I、II、III)的区域的插图,显示某些基因转录本的特征空间模式。Scale bar, 30 µm。
b. 激素相关基因(绿色)和受体相关基因(红色)的空间分布。当每个细胞中对应基因的计数大于 10 时,整个细胞用对应基因的颜色来标记。
c. 不同颜色的 top 空间基因共表达模块的热图。上面的颜色条表示共表达模块。右侧颜色条表示皮尔逊相关系数 (r) 值。
d. HDB 中不同基因的空间距离分析。与基因对 Snap25 和 Scg2 之间的空间共表达高度相关。
e. 基因对 Snap25 和 Scg2 与横轴列出的前 50 个表达基因的空间距离。平均最小距离是指从 Snap25 或 Scg2 基因转录本的信号点到前 50 个表达基因中每个基因的最近信号点的平均最小距离(通过平均像素测量)。Y 轴的单位是像素。
f. HDB 中六个聚类细胞的空间分布。
g. 散点图显示了 cluster 0 和 cluster 2 或 4 之间的空间轨迹推断。两条线和方向表示不同的分化轨迹。数字表示子类。
h. 细胞类群间配体和受体的原位空间相互作用的可视化。线条表示具有配体-受体相互作用的潜在细胞对。 d 和 f–h 中 X 轴和 Y 轴的单位是像素。对小鼠 HDB 中 100 个基因表达的检测重复至少 3 次。
根据空间转录组图谱,HDB 中鉴定的 100 个基因被分为 6 个 clusters(clusters 0 to 5),我们观察到这 6 个 clusters 沿着背腹轴显示出特定的空间分布模式(Fig. 3f and Extended Data Fig. 6f,g)。空间拟时序分析揭示了从 cluster 0 到 clusters 2 和 4 的时间转变(Fig. 3g)。由于 cluster 0 的细胞根据其空间分布明显聚集在一起,因此我们通过细胞轨迹推断来表征这些细胞与其他 clusters 之间的关系。通过结合空间信息和基因表达谱,这六个 clusters 进一步分为 131 个子类(0-130),拟时序-空间分析显示 cluster 0(subclass 6)中的两个分支与 cluster 2(subclasses 95 和 110)和 cluster 4(subclasses 65 和 90)相连。研究 HDB 中每个细胞 cluster 的发育和相关功能将非常有趣,因为这些细胞 clusters 在不同的位置被优雅地组织起来。
为了进一步探索 HDB 中详细的空间细胞-细胞相互作用网络,我们进行了详细的分析,将细胞分类为更多的 subclusters(Supplementary Fig. 9)。考虑到 HDB 中细胞 clusters 的空间定位,我们构建了一个空间细胞-细胞共定位网络(Supplementary Fig. 10a–d)。观察到 Ni4 subcluster 中少突胶质细胞和抑制性神经元之间的高频共定位(Supplementary Fig. 10d)。基于空间共定位和配体-受体基因表达信息的空间细胞-细胞相互作用分析揭示了某些相邻细胞 clusters 之间特定的配体-受体相互作用模式(Fig. 3h)。例如,我们观察到 Sst 在抑制性神经元 subcluster Ni5 中高表达,Sstr4 在相邻兴奋性神经元 subcluster Ne1 中高表达,Tac1 在抑制性神经元 subcluster Ni5 中高表达,Tacr3 在相邻兴奋性神经元 subcluster 2 (Ne2) 中高表达(Supplementary Fig. 10e)。这种空间细胞-细胞相互作用分析提示了神经元 subclusters 之间可能存在通信,例如 SST Ni5 与 Sstr4 Ne1 以及 Tac1 Ni5 与 Tacr3 Ne2。总之,我们的原位测序结果提供了 HDB 中详细的空间转录组,这可能为在单细胞空间水平上理解 HDB 的生理学铺平道路。
在高通量、高效描绘多重基因空间图谱的基础上,MiP-seq 被进一步用于探测大脑响应病原体感染的空间转录组变化。为此,我们使用 Extended Data Fig. 5 中相同的工作流程描绘了 PVN 响应结核分枝杆菌(M.tb)感染的空间转录组变化。217 个基因的空间转录组图谱,包括细胞标记、激素、受体和免疫相关基因,通过三轮测序在单细胞水平上被解码(Extended Data Fig. 7a)。空间转录组分析揭示了 M.tb 感染后 PVN 中几个免疫相关基因(Ly6h、Ccl3 和 Bcl6b)和受体基因(Gpr165、Calcr 和 Gria1)的上调(Extended Data Fig. 7b and data shown in Supplementary Table 3)。同时,M.tb 感染后,PVN 中的基因 C1qb、Pgm2l1 和 Ptgs2 下调(Extended Data Fig. 7b and data shown in Supplementary Table 3)。此外,我们根据 PVN 中的信号密度定量分析了某些细胞类型中的差异表达基因(DEGs),并获得了 M.tb 感染后单个内皮细胞、星形胶质细胞、小胶质细胞和神经元中的 DEGs(Extended Data Fig. 7c,d)。考虑到 PVN 在调节自主神经和神经免疫稳态中的关键作用,进一步研究这些空间 DEGs 在神经免疫调节中的作用将是非常有趣的。
4. 通过 MiP-seq 检测 DNA、RNA、蛋白质和神经递质
DNA 在细胞核中优雅地折叠和组织,3D 基因组架构协调遗传密码脚本的转录。因此,我们应用 MiP-seq 来破译大脑切片中 DNA 的空间组织(Fig. 4a)。为此,在 DNA 变性后,首先将锁式探针与起始引物一起与 DNA 靶标杂交,并使用 T4 DNA 连接酶进行连接。如 Fig. 4b 和 Extended Data Fig. 8a–c 所示,MiP-seq 清楚地揭示了 Rbfox3 和 Nr4a1 基因的空间定位。此外,我们利用 MiP-seq 检测 C6 胶质瘤中的 DNA 点突变(Fig. 4c)。我们的数据明确显示了 WT 和突变的 Nptxr 基因及其在神经胶质瘤细胞中的空间定位(Fig. 4c and Extended Data Fig. 8d–f)。
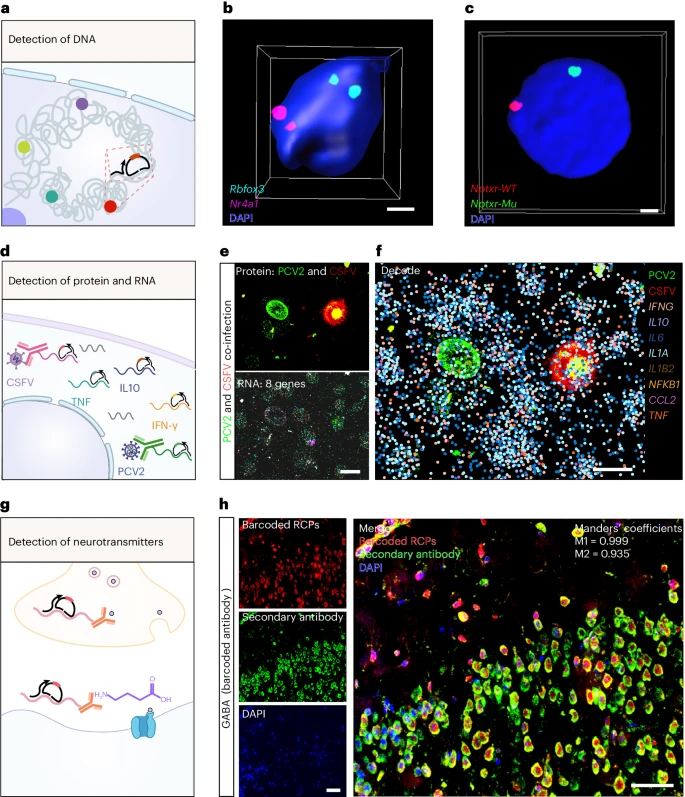
a. 通过 MiP-seq 对多个基因组位点进行原位共检测的示意图。
b. 利用 MiP-seq 原位检测小鼠脑切片中位于细胞核不同染色体上的 Rbfox3 和 Nr4a1 基因位点。
c. 位于 C6 细胞不同染色体上的 Nptxr 点突变的原位测序。
d. 同时原位检测多种蛋白质和 RNAs 的示意图。
e-f. MiP-seq 同时检测 PCV2 和 CSFV 共感染的 PK-15 细胞中 8 个细胞因子或趋化因子基因和 2 个病毒特异性蛋白(CSFV 的 E2 和 PCV2 的 Cap)的 mRNAs。
g. 基于核酸偶联抗体的 MiP-seq 原位检测 GABA 神经递质的示意图。
h. 在小鼠大脑皮层区域观察到通过 MiP-seq 与寡核苷酸偶联抗体(Cy3 红色荧光)检测到的 GABA 信号与通过经典免疫染色(Alexa Fluor 488 绿色荧光)检测到的信号相同。图像的共定位比表示为曼德系数。脑切片用 DAPI 复染色。a-h 中这些基因的检测至少重复了 3 次。Scale bars, 2 µm in b and c, 20 µm in e and f, 60 µm in h。
此外,MiP-seq 用于通过抗体-偶联核酸的原位测序来原位检测蛋白质。我们的数据证明,通过 MiP-seq 可以准确检测猪肾 (PK-15) 细胞中的 RNA 聚合酶 II (Pol II) 和小鼠大脑中的食欲素,并且使用显示相同基因表达的相同细胞通过传统免疫染色验证了这种特异性模式(Supplementary Fig. 11 and Extended Data Fig. 9a)。值得注意的是,由于通过 RCA 实现了强信号放大,与二抗免疫染色相比,MiP-seq 显示出更高的灵敏度(Supplementary Fig. 11 and Extended Data Fig. 9a)。此外,我们在 PCV2 和 CSFV 共感染的 PK-15 细胞中同时检测到四种蛋白,包括带有 Flag 标记的干扰素 γ、Pol II、猪圆环病毒 2 (PCV2) Cap 蛋白和猪瘟病毒 (CSFV) E2 蛋白(Extended Data Fig. 9b),表明 MiP-seq 用于原位蛋白质检测的高通量能力。此外,我们通过 MiP-seq 同时检测蛋白质和 mRNAs,以探索 8 个细胞因子/趋化因子基因响应 PCV2 和 CSFV 病毒感染的动态表达(Fig. 4d–f and Supplementary Fig. 12)。此外,我们还通过 MiP-seq 同时共同检测 PCV2 基因组 DNA、PCV2 Cap 蛋白和 8 个细胞因子/趋化因子基因的 mRNAs,证明了 PK-15 细胞对 PCV2 感染的反应(Supplementary Fig. 13)。
由于蛋白质检测转移到与其特异性抗体偶联的核苷酸序列的检测,因此 MiP-seq 每轮测序可以检测到 10 个蛋白质信号。因此,理论上,如果可以获得高质量的 DNA 偶联抗体,蛋白质检测也可以扩展到 。为了进一步证实 MiP-seq 蛋白检测的高通量能力,我们对人弥漫性大 B 细胞淋巴瘤 (DLBCL) 组织切片中的 30 种免疫相关蛋白和 6 个诊断标记基因 mRNA 进行了联合检测(Extended Data Fig. 10)。在相同的 DLBCL 切片中以单细胞分辨率解码了 30 种蛋白质和 6 个基因的 mRNAs 的空间表达模式,其中细胞分为 16 个 subclusters(Extended Data Fig. 10a,b)。随后,描绘了这些细胞及其细胞间通讯网络的空间图,其中展示了抗原呈递细胞-2(APC-2)(HLA-DR )、活化的 T 细胞(CD134 )和淋巴瘤-2 (CCND2 ) 细胞(Extended Data Fig. 10c–e)。有趣的是,我们观察到中央记忆细胞(CD27 /CD45RA )和终末分化效应记忆细胞(CD27 /CD45RA )之间存在明显的空间分布模式(Extended Data Fig. 10f,g)。值得注意的是,我们的数据显示趋化因子和趋化因子受体 CCR7 和 CXCR6 与 CD45RA 具有相似的表达模式,这可能与效应记忆细胞的功能有关(Extended Data Fig. 10f,g)。
神经递质和激素等生物分子的精确时空分布在细胞通讯和激活中发挥着关键作用。非常需要一种高通量、高灵敏度的检测方法,能够实现生物分子的亚细胞分辨率。由于许多生物分子具有半抗原特征并且可以被抗体特异性检测,因此我们应用 MiP-seq 通过抗体偶联核酸的原位测序来探索神经递质的时空分布(Fig. 4g)。为此,我们使用抑制性神经递质 γ-氨基丁酸(GABA)作为脑切片中检测的生物分子。如 Fig. 4h 所示,MiP-seq 清楚地揭示了 GABA 的空间分布,并且该结果通过传统免疫染色得到了验证。总之,这些数据表明,多功能 MiP-seq 策略可用于检测不同的生物分子,包括 DNAs、RNAs、蛋白质、神经递质和激素。
5. 多重成像分析与 MiP-seq 检测的空间基因图谱相结合
单个细胞的多样性和特定功能高度依赖于它们的基因谱。在中枢神经系统中,邻近的神经元可以表现出不同的功能活动和一系列基因表达。通过开发一种体内原位方法,在同一动物中结合双光子 Ca2+ 成像和 MiP-seq,我们能够将同一单个神经元的功能活动和基因表达谱联系起来。简而言之,首先在 Thy1-GCaMP6s 小鼠的初级视觉皮层中进行体内双光子 Ca2+ 成像,以识别来自 ~350 × 350 × 80 μm3 体积中所有神经元的尖峰相关胞体钙信号。随后,对具有不同神经元标记基因和神经递质基因(如 Gad1、Vip 和 NPY)的相应成像区域的切片进行 MiP-seq。原位 MiP-seq 图像中的核信号用于对齐体内双光子 Ca2+ 图像堆(Fig. 5a–c, and Supplementary Figs. 14 and 15a,b)。对于每个识别的神经元,分别从 Ca2+ 图像和 MiP-seq 数据集中提取不同的 Ca2+ 活性和基因表达谱(Fig. 5d and Supplementary Fig. 15c,d)。在 Ca2+成像和 MiP-seq 相结合的基础上,我们观察到 Ca2+ 瞬时峰值幅度较高的细胞显示出较高的 Nov 表达,而 Ca2+ 瞬时平均幅度较高的细胞具有较高的 Slc17a7 表达(Fig. 5e,f) 。
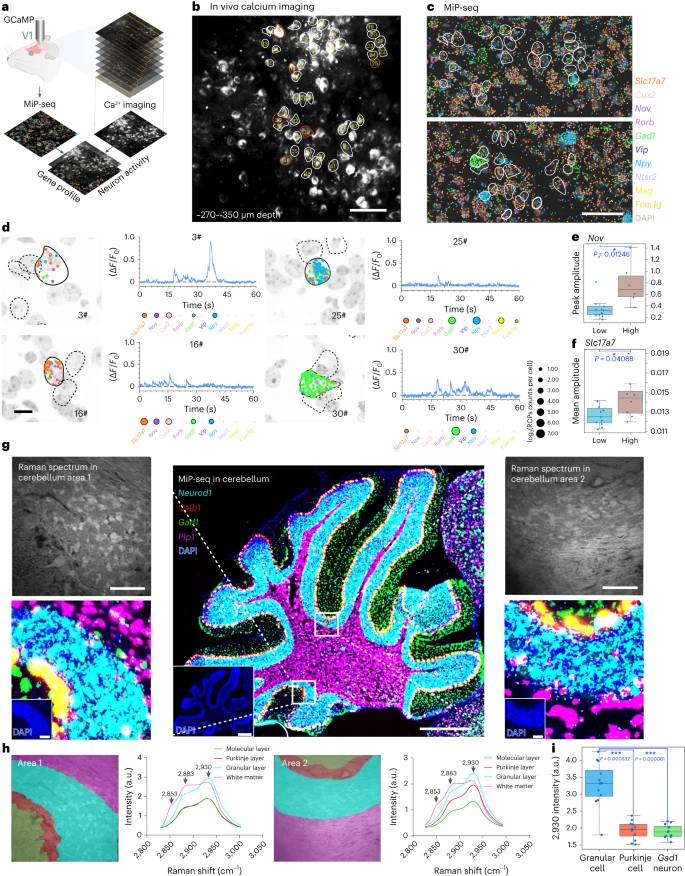
a. 整合体内钙成像和 MiP-seq 的原位多组学示意图,用于将小鼠视觉皮层的瞬时生理状态和原位基因表达谱联系起来。
b. 视觉皮层体内钙成像(-270 ~ -350 μm深度)。用圆圈标记的细胞被编号(1 至 45)并通过 MiP-seq 进行原位基因表达谱分析。
c. 在对应于钙成像区域的同一小鼠视觉皮层的脑切片中共同检测 10 个标记基因转录本。
d. MiP-seq 在几个细胞中检测到的标记基因表达的呈现,显示出不同的钙动态。实心外壳表示用于呈现标记基因表达和钙动态的细胞。虚线框表示其他分析的细胞,没有呈现标记基因表达和钙动态。
e-f. Nov (e) 和 Slc17a7 (f) 表达水平较高的细胞分别表现出较高的 Ca2+ 瞬时峰值和平均幅度。根据标记基因的中值表达水平将细胞分为高表达组和低表达组。使用 Mann-Whitney U test (two-tailed) 确定统计显着性,*P < 0.05,n = 19 个细胞,用于 Nov 和 Slc17a7 基因表达与 Ca2+ 瞬时幅度的关联分析。数据以四分位距 (IQR) 形式呈现。
g. 整合高光谱 SRS 和 MiP-seq 的原位多组学。拉曼成像后,通过 MiP-seq 在小鼠大脑的相同小脑切片(矢状面,50 µm 厚度)上检测四种细胞标记基因(Neurod1、Calb1、Gad1、Plp1)的表达。中间面板中的方框区域 1 和 2 分别在左侧面板和右侧面板上放大。
h. g 中方框区域 1 和 2 的不同小脑层的高光谱 SRS。
i. 与浦肯野细胞和抑制性分子层中间神经元相比,颗粒细胞的 2,930 波长强度更高。使用 Kolmogorov–Smirnov test (two-tailed) 确定统计显着性,***P < 0.001,n = 12 cells per group。数据以 IQR 形式呈现。在 e、f 和 i 的箱线图中,水平线表示中值,晶须从最小值延伸到最大值。具体来说,上晶须延伸到不超过四分之三分位数 + 1.5 × IQR 的最后一个数据点,而下晶须延伸到不小于四分之一分位数 - 1.5 × IQR 的最后一个数据点。e、f 和 i 的原始数据可在 Supplementary Table 3 中找到。使用 MiP-seq 进行体内钙成像的联合检测以及使用 MiP-seq 进行受激拉曼散射的联合检测重复至少 3 次。Scale bars,b 和 c 中为 50 μm,d 中为 10 μm,g 中为 500 μm(较低放大倍数)和 50 μm(较高放大倍数)。
此外,我们将 MiP-seq 与高光谱受激拉曼散射成像相结合,以协调生化分子图谱与原位基因图谱。通过 MiP-seq 在小鼠大脑的小脑和穹窿下器官中检测到细胞标记基因的原位表达,并分别与相同和最接近的拉曼光谱图像对齐(Fig. 5g–i and Supplementary Fig. 16)。具体的拉曼光谱和相关的基因表达模式如 Fig. 5h,i 和 Supplementary Fig. 16 所示。我们的数据表明,不同的小脑细胞层显示出不同的拉曼光谱,并且与小脑细胞相比,颗粒细胞具有显着更高的 2,930 波长强度。浦肯野细胞和抑制分子层中间神经元(Fig. 5h,i)。我们还将血管结构图像与 MiP-seq 数据结合起来,同时观察了 Gad1 基因的表达和小鼠大脑弓状核中的微血管结构(Supplementary Fig. 17)。
6. 解决光学拥挤信号的顺序稀释策略
在多轮原位高通量解码过程中,光信号拥挤,尤其是高表达水平基因,是信号查询中最具挑战性的步骤。为了克服这个问题,我们开发了一种连续稀释 MiP-seq 策略,其中将基因分配给具有不同独特稀释引物序列的不同条形码组。这样,通过使用不同的测序引物,信号在各轮成像中被稀释。值得注意的是,我们选择了某些基因作为参考基因,每轮都通过锚定引物对这些基因进行测序(Fig. 6a)。经过多轮测序和成像,所有基因信号在参考基因信号比对的基础上叠加,重建高密度空间转录组图谱。作为概念证明,使用参考基因 Gfap 和 Gad2 进行比对,对 Cxcl2、Agtr1b、Slc17a6、Ccl2、Il1b、Col1a1、Tnf 和 Calcr 进行两轮连续稀释 MiP-seq。如 Fig. 6b–d 所示,我们的策略确实可以实现精确的原位基因信号稀释和表达模式重建。因此,这种连续稀释 MiP-seq 方法可以极大地促进高通量转录组绘制过程中高密度信号的精确查询。
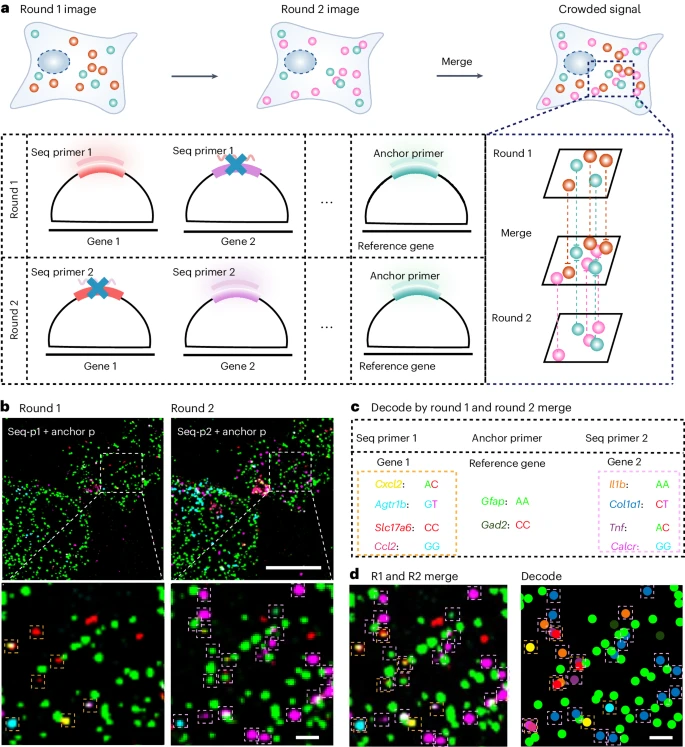
a. 顺序稀释策略示意图。单细胞中高度拥挤的基因信号被稀释到多轮中。基准基因由每轮测序的锚定引物查询,而其余基因则分为多轮并由不同组的测序引物测序。在基准基因信号注册的基础上,重建来自不同测序轮次的其余基因的信号,以实现单细胞中的拥挤信号查询。
b-d. 通过对小鼠皮层培养神经元进行两轮 MiP-seq,Cxcl2、Agtr1b、Slc17a6 和 Ccl2(第 1 轮)以及 Il1b、Col1a1、Tnf 和 Calcr(第 2 轮)的 mRNA 信号被重建,在基准基因 Gad2 和 Gfap 信号注册的基础上。通过连续稀释策略检测拥挤信号至少重复三次。Scale bar,20 μm(b 中的低放大倍数)、2 μm(b 中的较高放大倍数)和 d。
讨论
我们开发了一种高通量 MiP-seq 策略,以亚细胞分辨率有效描绘组织中的多重 DNAs、RNAs、蛋白质和神经递质图谱。与当前的原位测序方法相比,MiP-seq 利用双条形码锁式探针和成对测序策略,与其他方法相比大大提高了解码能力( vs )并且需要更少的测序轮次。因此,MiP-seq 可以通过将测序时间缩短约 50% 来降低测序和成像成本。此外,双条形码和成对测序策略大大减少了成像时间,从而最大限度地减少了激光损伤,这是原位测序过程中的一个关键问题。此外,与当前的原位测序方法相比,我们设计的起始引物和锁式探针提高了信号特异性和功效。与 MERFISH 和 SeqFISH 等高通量 FISH 技术相比,MiP-seq 可以检测短 RNAs(小于 500bp),并且可以区分单碱基对突变,同时需要更少的信号解码周期。虽然 MiP-seq 具有多种优势,但它需要 Z 轴扫描来精确对齐多轮测序之间的信号点,这需要更长的信号解码时间,尤其是对于大视野。
由于 MiP-seq 可以实现单核苷酸分辨率,我们将其应用于检测肿瘤基因突变和亲本基因的等位基因特异性表达。由于该方法可以同时检测完整病理切片中的基因拷贝数变异和基因突变,因此可用于精确的肿瘤诊断。检测亲本基因的等位基因特异性表达可以极大地提高对许多基本生物学现象的理解,例如杂种优势、显性遗传和基因上位性。值得注意的是,我们的数据表明 MiP-seq 对 RNA m6A 修饰敏感,因此有可能区分 m6A 修饰的 RNA 和未甲基化的 RNA。未来,开发一种类似 DNA 亚硫酸氢盐开关的技术,以化学方式用另一种碱基替换 m6A 修饰的碱基,这将很有趣,这将能够使用 MiP-seq 原位精确检测 m6A 或其他 RNA 修饰。
基于 MiP-seq 的高效率,仅在 2-3 个测序轮次内即可同时检测到数百个基因,这使我们能够绘制 HDB 中的空间图谱并探索 M.tb 感染引起的 PVN 中空间转录组改变。这些结果例证了使用 MiP-seq 描绘某些脑核的空间组学以及识别某些位置和特定细胞类型的空间组学变化。根据 MiP-seq 识别的 HDB 空间分布,可以识别多种细胞间相互作用,例如自分泌、旁分泌以及激素或神经递质之间的长程相互作用。进一步破译神经系统中的长程相互作用,特别是基于环路的长程相互作用非常重要。在这种情况下,需要将神经环路示踪技术与配体-受体基因表达数据的原位检测相结合。此外,在 DLBCL 组织切片中共同检测 30 种免疫相关蛋白,支持了 MiP-seq 空间蛋白质组学的高通量能力。更重要的是,MiP-seq 的蛋白质检测不再因二抗问题而受到抗体来源物种的限制,这意味着来自同一物种的抗体可以同时用于多重蛋白质检测。
此外,我们将 MiP-seq 与体内 Ca2+ 成像和拉曼成像相结合,获得了具有多维信息的组织图谱,这对于将细胞表型和功能与基因表达谱联系起来非常有用。MiP-seq 还可以与其他成像技术相结合,例如 pH 成像、氧化应激成像和电活性成像。尽管我们在研究的现阶段没有直接确定基因表达模式与特定钙动力学和拉曼光谱之间的关系,但我们的方法提供了概念证明,以探索动态基因表达谱与不同细胞功能之间的对应关系以及原位、单细胞和高通量水平的表型。随着深度学习的进步以及整合原位基因型和不同类型表型数据(包括时空生物分子信息和钙活性)数量的增加,该方法可以为通过原位基因图谱预测细胞的拉曼光谱、形态和功能铺平道路,反之亦然。未来,我们或许可以通过拉曼成像来预测准确的肿瘤边界,这可能有助于手术中肿瘤的精确切除。
此外,我们基于 MiP-seq 进行了原位多组学分析,以亚细胞分辨率描绘 DNAs、RNAs、蛋白质和小生物分子(神经递质)的空间景观。考虑到其高通量检测能力,MiP-seq 可用于通过多轮测序全面解读 3D 基因组景观,这将增强对 3D 基因组中协调基因转录的理解。对于蛋白质和生物分子的原位检测,可以使用核酸适配体,因为它们可以被设计为特异性结合蛋白质/生物分子,并且可以轻松地与核酸偶联(Supplementary Fig. 18)。此外,由于光信号拥挤导致几乎所有基于成像的空间组学方法的“瓶颈”,我们开发了一种连续稀释策略,使用一组测序引物和锚定基因引物来解决这个问题。这种稀释策略将有助于精确询问高密度信号,例如高表达基因的信号。
总体而言,MiP-seq 是一种仅需 N 轮测序即可对 10N 个基因进行空间分析的有效方法,具有高通量、单核苷酸和多组学检测功能。它可以与功能成像相结合,获得具有多维信息的组织图谱。许多相关技术可以得到实质性改进,包括超分辨率和低毒性成像系统、长读长测序策略以及功能成像与原位代谢组质谱的整合。这些技术的进一步发展可能允许对许多组织和器官的详细综合分子和功能图进行多维重建。
注:本文为个人学习笔记,仅供大家参考学习,不得用于任何商业目的。如有侵权,请联系作者删除。

本文由 mdnice 多平台发布