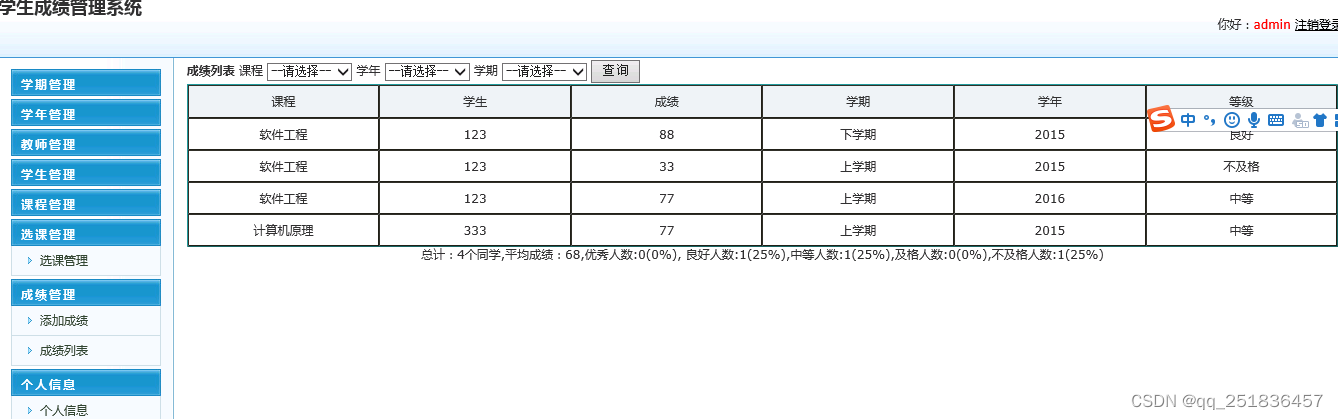
PacBio研究专题
二代测序读长偏短,环境宏基因组样品研究受到了很大限制。作者通过三代测序对来自地中海的冬季混合海水样本进行宏基因组测序。利用PacBio Sequel II平台的超长读长明显可以提升宏基因组的组装质量,又能显著提升MAGs质量。研究人员通过比较PacBio Sequel II平台CCS(HiFi)模式及传统的illumina NGS的组装指标,证实了上述结论:
研究者利用SPAdes 3.14、Flye v2.7和HiCanu v2.0等不同软件组装CCS数据,同时与NGS常规流程进行比较:PacBio组装结果的contig N50可达到5.35-5.95 Mb;是NGS的几十倍,其中最长的contig达到了2.6Mb,正好是NGS的十倍。从基因的平均长度来看,PacBio提供了更可靠的结果。
下表为短读长(SR)与长读长(LR)的数据比较:

基于PacBio数据的MAGs构建
相比于NGS筛选2K contigs进行MAGs的生成,PacBio的数据可以从容挑选5K以上的序列开展分析。三代测序获得的中高质量MAGs(>50%完整性,且污染率<5%)与NGS测序结果比较发现,除了数量上的提升外,基于三代测序获得的MAGs的contig的平均长度提高了4倍,且最长的contig提高了2.7倍,重要的是基因组碱基准确度得到了明显提升,在同等成本下基因组组装消耗的计算资源和时间大幅下降。
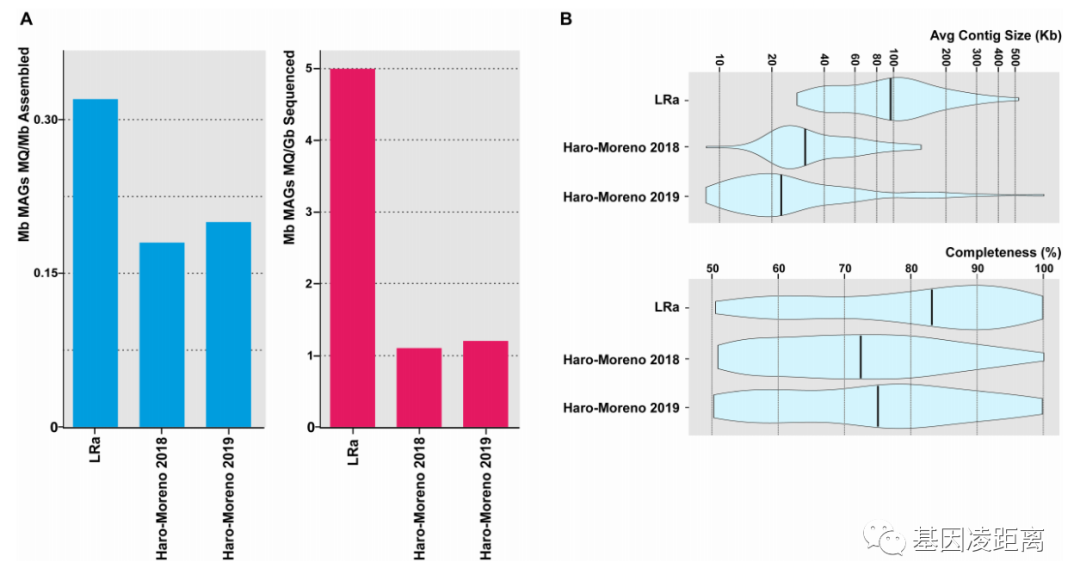
(A) 本研究(LR)中中等质量MAGs(>50%完整,<5%污染)与NGS结果的比较(分别为蓝色柱图与红色柱图)。
(B) 小提琴图显示了窗格中描述的MAGs的平均连续大小和完整程度.
结论:
1) 利用三代测序的long reads可以直接获得完整的基因,避免基因组装造成的误差。
2) 三代测序的HiFi reads有助于重建更完整的元基因组(MAGs),特别是针对具有高度多样性的微生物。
3) 利用三代测序重建的MAGs完整性更高,包含更多自适应基因,可用于生物技术潜能挖掘。
4) 基于PacBio测序的CCS模式获得的序列,错误率更小,更适用于针对复杂微生态样本的MAGs研究。
doi:10.3389/fmicb.2021.708782
拥有最新的三代测序PacBio技术平台,可以为广大科研用户提供多种三代测序与分析技术服务。截至目前,已开展并完成几十例宏基因组HiFi测序及组装项目,拥有丰富的宏基因组三代测序组装经验。