百趣代谢组学文献分享,自2014年瑞士苏黎世联邦理工学院的Picotti和她的研究小组开始用Lip-SRM法测量复杂蛋白质混合物的大量结构改性蛋白质以来[1];该研究小组随后对方法进行改进,研究了复杂细胞基质中几种生物蛋白质的热稳定性,并推翻长期以来关于蛋白变性的定论,提出蛋白质变性全新理论[2];该研究小组又以该技术为基础发现细菌细胞中的蛋白和小分子代谢之间之前未知的相互作用。
- 代谢组学文献分享—前言
生物体的各项生命活动以体内分子间的相互作用为基础,如蛋白合成过程中,蛋白和DNA间、蛋白和蛋白间以及蛋白和RNA间的相互作用[3];除此之外,小分子代谢物对于许多蛋白来说是非常重要的调节因子,在信号转导、新陈代谢和蛋白翻译过程中起着重要作用。但是,人们一直致力于研究蛋白与生物大分子间的相互作用,对蛋白质与小分子物质间相互作用的研究相对滞后。代谢组学文献分享,随着近几年质谱仪、高通量蛋白组学和代谢组学的兴起,一项涉及生物分子之间的相互作用的新组学-相互作用组学(interactomics)也进一步发展,这也预示着生物医学研究和医药领域的改变[4]。
目前,对蛋白和代谢物间的相互作用主要基于一些特定类型(如催化)的相互作用或检测小蛋白片段与代谢物间的相互作用,需要对代谢物进行化学修饰或选取某些特定类型的化合物。代谢组学文献分享,因此,Paola Picotti课题组利用代谢物与蛋白结合会引起蛋白质结构改变的特点,开发了新的Limited (or native) proteolysis-small molecule mapping(LiP-SMap)法用于研究小分子代谢物和蛋白之间的相互作用,并开始在全蛋白组水平上系统地分析和定量了蛋白与代谢物间的相互作用,并建立了它们之间的关系。
2.代谢组学文献分享—LiP-SMap法技术路线与方法验证
如图1,与之前的Lip-SRM法[1,5]类似,首先在非变性的条件下提取蛋白质,然后将提取的蛋白分为两组,一组加入小分子代谢物,让其与蛋白质相互作用,另一组不进行任何处理;再加入广谱的蛋白酶K(PK)产生结构特异性蛋白片段;得到的特异片段再经序列特异性的Trypsin酶解,最后用LC-MS法定量得到的肽段。代谢组学文献分享,由于代谢物与蛋白的特异性结合会引起蛋白结构的局部改变,从而改变蛋白质被PK酶切的表位产生不同的肽组,通过与对照组的比较筛选出代谢物与蛋白的作用图谱,从而找出结合位点。
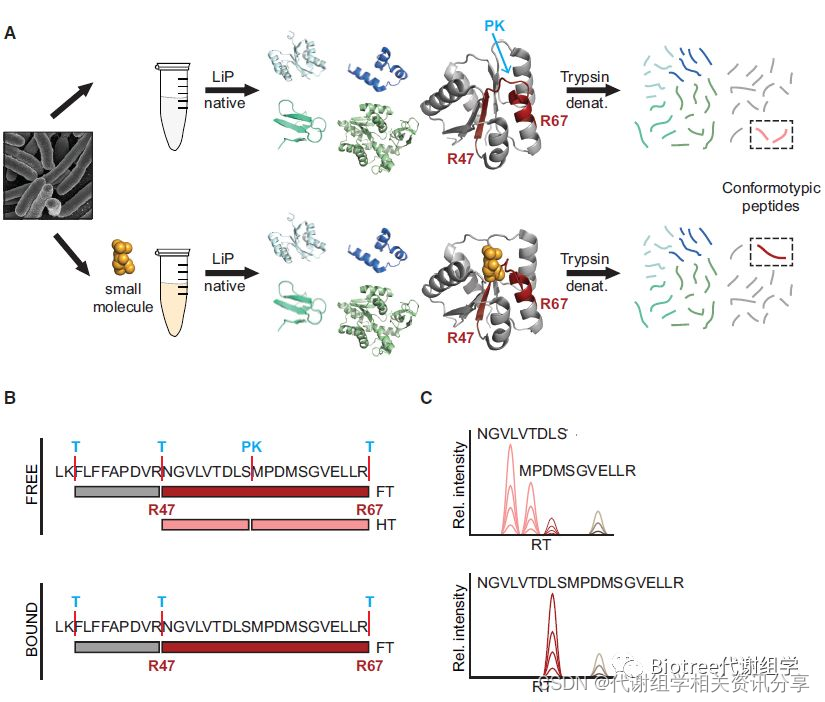
图1 LiP-SMap法技术路线(A:蛋白提取与酶解; B:肽段序列; C:MS检测的肽段信号)
为验证该技术的可靠性,选取有已知蛋白与代谢物作用位点的腺苷三磷酸(ATP)、苯丙氨酸(L-Phe)和磷酸烯醇式苯酮酸(PEP)3个物质进行研究,并加入E.coli的蛋白提取液中;代谢组学文献分享,还用该方法筛选酵母细胞中与浅蓝菌素结合的位点Fas2进行实验。
3. 代谢组学文献分享—大肠杆菌中的代谢物-蛋白质
互作网络和结合位点分析
选择E.coli作为研究对象,分别使用20种(图2A,8个中心碳代谢相关代谢物,4个氨基酸,7个核苷磷酸和1个环腺苷单磷酸)不同的代谢中间产物检测其与大肠杆菌蛋白的相互作用。代谢组学文献分享,通过实验,共检测到1,678个代谢物-蛋白质的相互作用,其中1,447个未被报道过(图2B)。进一步研究发现很多新相互作用与有机酸和糖磷酸盐相关。
此外,将检测到的代谢物-蛋白作用和BRENDA数据库对比,发现该检测方法的假阳性率不超过5.5%(有些新发现的作用由于未在数据库中,也被认为是假阳性结果)。代谢组学文献分享,为评估相互作用结果的可信度,该团队也开发了STAR法对相互作用进行打分(图2C)。
该团队通过测定Protein Data Bank Ligand Expo数据库中收集的代谢物蛋白复合物的结构,提供了基于高通量蛋白组水平的代谢物结合位点的谱图信息(图2D)。

图2.代谢物与蛋白相互作用图(A:中心碳代谢网络图; B:基于LiP-SMap检测出的作用位点; C:1,678个LiP-SMap相互作用位点的得分分布; D:以GTP为代表的结合位点结构)
4. 代谢组学文献分享—代谢物和蛋白相互作用特征与新调节方式发现
通过分析代谢物敏感的蛋白与E.coli的“核心蛋白组”之间的关系发现,代谢物更倾向于与浓度随时间的变化较小或保持不变的蛋白结合(图3A)。代谢组学文献分享,这也可能预示在细胞调节蛋白质活性过程中,代谢物-蛋白的结合与蛋白浓度的变化是两条互补的途径。
代谢物通过变构、催化或配体诱导等方式实现酶的活性调控,那么是不是还有新的作用方式呢?该团队又选取几个参与中心碳代谢的代谢物与酶(柠檬酸与磷酸烯醇式丙酮酸羧化酶Ppc,果糖-1,6-二磷酸FBP与磷酸烯醇式丙酮酸合酶调控蛋白PpsR, FBP与果糖-6-磷酸脱氢酶)的相互作用进行研究,发现代谢物通过间接的方式调控酶的变构从而调节酶的活性(图3B,C)。
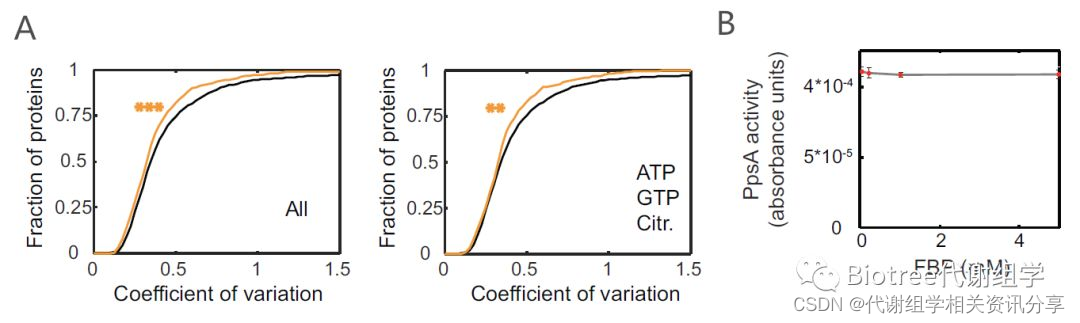
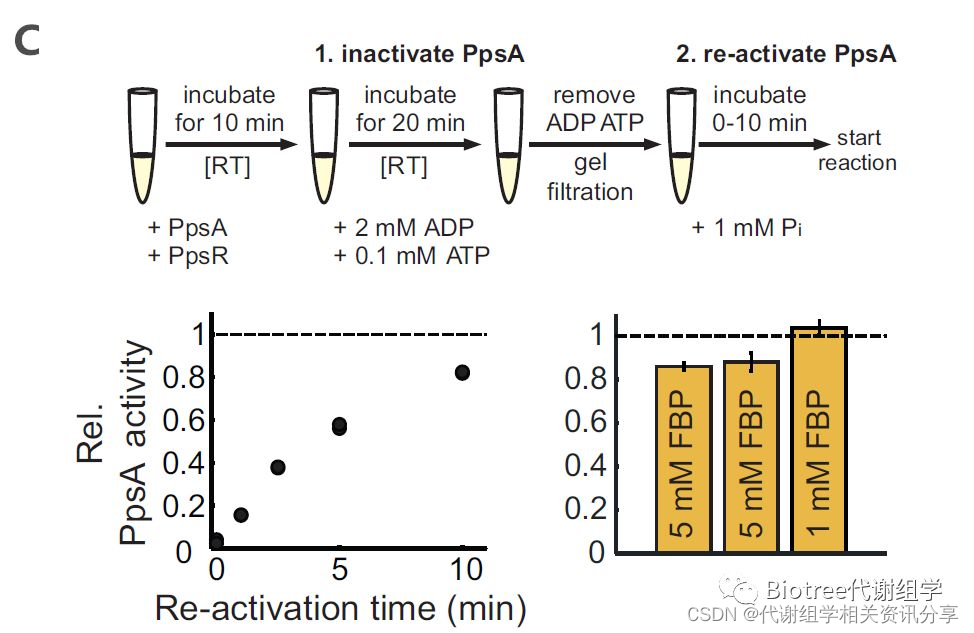
图3 代谢物和蛋白相互作用特征(A, 左:至少有一个代谢物结合位点的蛋白的累积变异系数曲线; 右: 排除与ATP、GTP和Citr.结合位点的蛋白的累积变异系数曲线; 橙:改性蛋白, 黑:非改性蛋白)与新调节方式发现(B,C: PpsA活性测定)
5. 代谢组学文献分享—代谢物与蛋白在催化位点的相互作用
为研究催化活性或竞争性抑制,该团队首先分析了大肠杆菌蛋白晶体结构信息,发现活性位点中代谢物与异构肽段的距离为6.44埃(图3A),并将6.44埃认为是活性位点作用范围,大于6.44埃的远端位点可能是变构位点或其他催化位点(图3B,C)。
接着,对1,665个异构肽段的分析,发现37%的肽段是与已知化合物结合的活性位点,其中有部分位点理论上能与催化中心形成氢键(图3D)。代谢组学文献分享,此外,也发现有多个代谢物结合位点的酶大都具有氧化脱氢酶活性或参与涉及磷酸转移的反应(图3E)。
此外,该团队也通过实验进一步证明,LiP-SMap法能用于鉴定新的酶-底物相互作用和竞争性抑制位点。
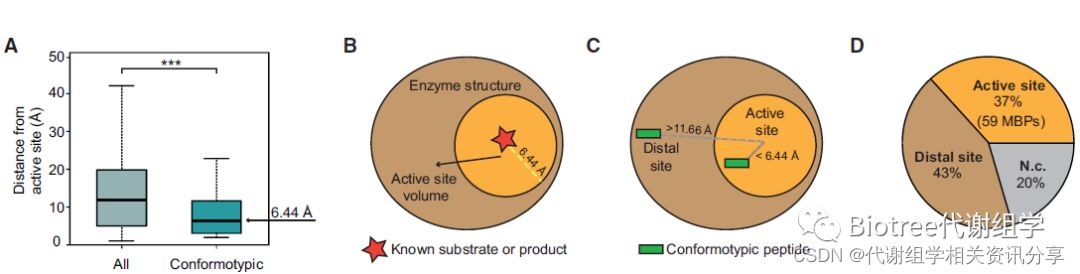

图4 代谢物与蛋白质结合位点的分析(A: 酶-代谢物复合物结构活性位点中所有检测到的肽段与变构肽段的最小距离分布; B and C: 活性位点分布; D: 活性位点和远端位点分布; E: 代谢物与酶结合位点分布,绿色-结合位点已知)
6. 代谢组学文献分享—代谢物结合对蛋白高级结构的影响和量化指标
之前的研究中已经有学者提出代谢物的结合可引起蛋白复合物或者聚合物的形成或解离,该团队也选取162个蛋白-代谢物对进行分析,发现约68%的蛋白-代谢物对变得更加敏感,另外32%抗性增加;另外也发现并验证了代谢物的结合除引起结构重排、形成同系复合物或者作为杂合物亚基单元外,也能引起已知蛋白复合物的组成发生改变。
最后根据ATP结合位点可以对于ATP的结构反应度进行区分的特点,研究组提出了代谢物结合的量化指标in-extract Kds(图5);in-extract值在1-10mM范围内时,有83%蛋白-代谢物相互作用是新发现的;代谢组学文献分享,同时发现了111个新低亲和位点, in-extract平均值为3.23。这也再次证明LiP-SMap法可直接用于确定复杂生物样本中代谢物结合位点的量化指标。
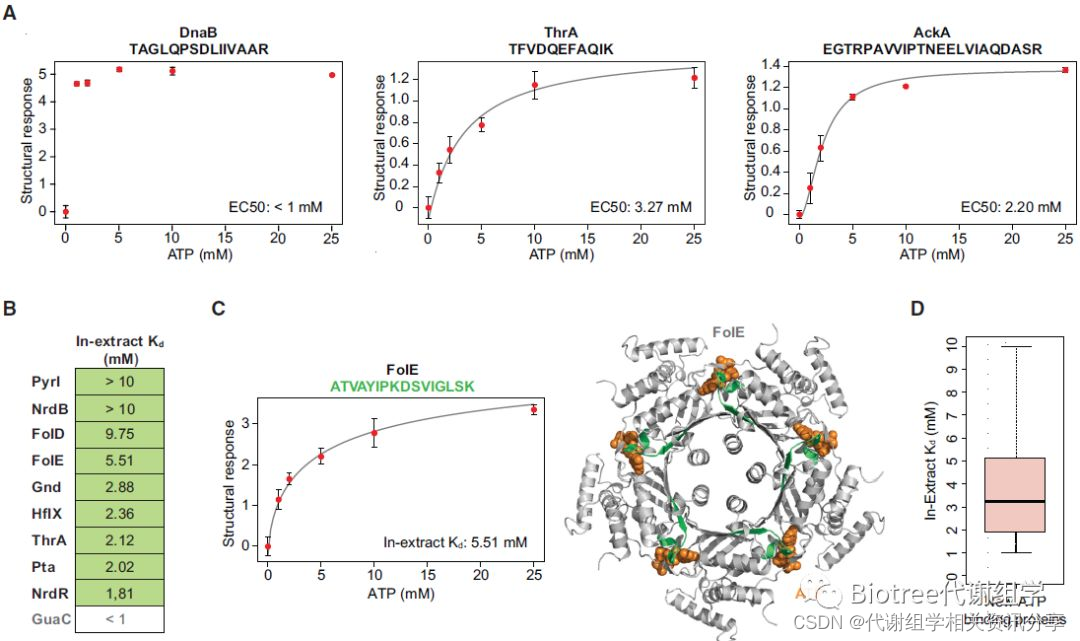
图5 LiP-SMap法测定细胞中代谢物结合位点亲和力
7. 代谢组学文献分享—小结
本文利用新开发的LiP-SMap法,结合生物化学实验、代谢组以及蛋白组方法创新性地研究了代谢物-蛋白间的相互作用,并验证了该方法可用于新的变构和催化位点的发现;同时也揭示代谢物与蛋白的结合会引起蛋白高级结构的变化。
代谢组学文献分享,该方法简单高效,不需要特殊的化学反应或者添加保护基团,可用来测定药物与细胞蛋白之间的相互作用并确定药物的作用靶标,为制药行业的研究提供了一种新的有效工具。
代谢组学文献分享—参考文献:
[1] Yuehan Feng, etal Global analysis of protein structural changes in complex proteomes. Nat Biotechnol.2014 Oct;32(10):1036-44.
[2] Leuenberger P , et al. Cell-wide analysis of Protein thermal unfolding reveals determinants of thermostability.Science.2017 Feb 24;355(6327).
[3] Tagore R, et al. A Global Metabolite Profiling Approach to Identify Protein-Metabolite Interactions. J Am Chem Soc.2008 Oct 29;130(43):14111-3.
[4] Guo H,Peng H,Emili A. Mass spectrometry methods to study protein-metabolite interactions. Expert Opin Drug Discov.2017 Dec;12(12):1271-1280.
[5] Schopper S, et al. Measuring protein structural changes on a proteome wide scale using limited proteolysis-coupled mass spectrometry. Nat Protoc.2017 Nov;12(11):2391-2410.