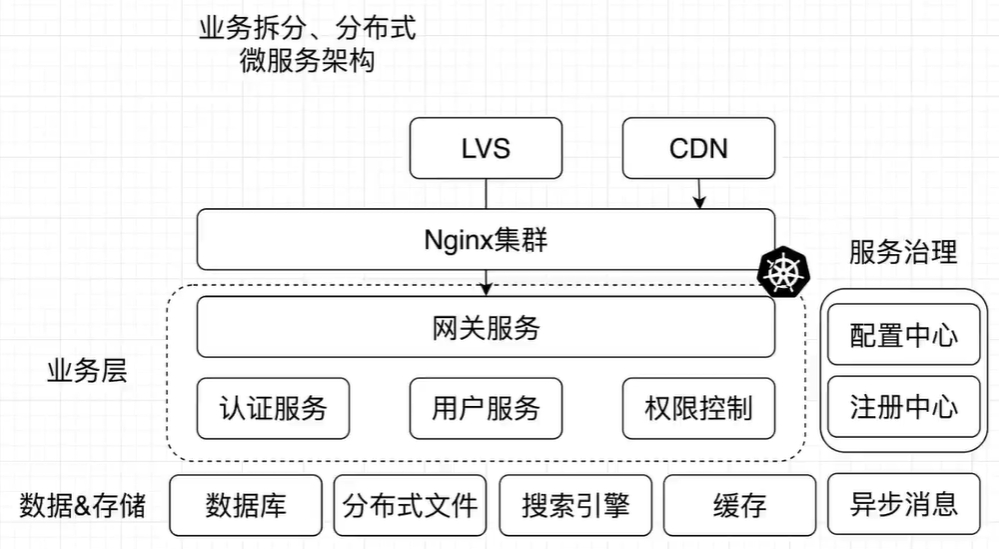
像GPT这样的LLM在自然语言任务上表现出了令人印象深刻的性能。这里介绍一种新的方法,通过将基因表达数据表示为文本,让这些预训练的模型直接适应生物背景,特别是单细胞转录组学。具体来说,Cell2Sentence将每个细胞的基因表达谱转换为按表达水平排序的基因名称序列。实验证明,这些基因序列,称之为“cell sentences-细胞句子”,可以用来微调GPT-2等语言模型。至关重要的是,作者发现自然语言预训练可以提高模型在细胞句子任务中的性能。当对细胞句子进行微调时,GPT-2在提示细胞类型时会生成生物学合理的细胞。相反,当提示使用细胞句子时,它也可以准确预测细胞类型标签。这表明,使用Cell2Sentence微调的语言模型可以获得对单细胞数据的生物学理解,同时保留其生成文本的能力。该方法使用现有的模型和库将自然语言和转录组学结合起来。
来自:Cell2Sentence: Teaching Large Language Models the Language of Biology, ICML, 2024

- 图1:Cell2Sentence框架概述。输入的单细胞数据(包括元数据)被转换为单细胞句子,用于语言模型微调。
- 在推理时,新的细胞句子被生成,并且可以被转换回基因表达空间。
目录
- 背景概述
- 相关工作
- 方法
- 数据转换
- 任务
- 实验
- 微调数据集
- 训练的相关内容
- 方式1:C2S预训练
- 方式2:NL+C2S微调
- 实验1:条件细胞生成
背景概述
将LLM应用于生物学等其他领域仍然是一个悬而未决的挑战。目前该领域的方法依然依赖于专门设计的神经网络,难以直接利用来自自然语言的LLM知识。
现在目标是将LLM的能力扩展到转录组学领域。关键思想是通过一种称为Cell2Sentence(C2S)的方法,以符合LLM的文本格式表示单细胞数据。C2S将每个细胞的基因表达谱转换为按表达水平排序的基因名称(也称为gene symbols)的序列。这允许任何预训练的因果语言模型在细胞序列上被进一步微调。其次,自然语言预训练结合C2S训练显著提高了转录组任务的模型性能,并且性能还随模型大小而扩展(scaling law)。我们不仅可以生成和解释转录组学数据,还可以用自然语言进行交互。
潜在的应用包括推断基因表达在扰动下的变化,生成罕见的细胞类型,识别marker,以及通过自然语言解释转录组学。这种能力可以协助我们推进单细胞研究。便利性包括两方面:
- Easy to use:任何用户都可以使用流行的第三方库(如Hugging Face Transformer)轻松利用任何可用的预训练语言模型,这些库具有简化的模型部署和微调功能。唯一需要的额外步骤是将原始单细胞数据预处理为C2S格式,并将生成的文本后处理回基因表达向量,这两个步骤都是使用GitHub存储库中提供的模块化数据转换管道无缝完成的。
- Easy to modify:训练管道的简单性意味着用户可以根据自己的喜好塑造预处理和训练步骤,例如通过添加更多元数据或特定提示。

- 图2:Cell2Sentence框架的详细概述。通过基因名称的表达排序,将单细胞基因表达谱转化为细胞句子。这些细胞句子可以用生物学元数据进行注释,包括细胞类型、组织或疾病。随后,使用细胞句子对语言模型进行微调。在推理中,细胞句子是有条件地(例如,给定细胞类型或组织类型)和无条件地生成的。生成的细胞句子可以转换回基因表达谱。
相关工作
LLM
LLM已适用于自然语言处理领域中关于文本数据的广泛任务。一些关键任务和示例架构包括文本分类(LSTM、BERT、RoBERTa)、问答(LlaMA-2、Falcon)和文本生成(T5、GPT-3、BART)。
单细胞基础模型
目前已经存在了几个任务的体系结构,包括:细胞注释–根据细胞的生物学特性为其分配标签(ACTINN、scVI),批次整合–去除技术引起的转录物丰度差异(scVI、scGen、SAUCIE),以及插补–推断缺失的转录物丰度数据(scVI,DeepImpute、SAUCIE)。最近,GEO和人类细胞图谱(HCA)等努力集中并标准化了数百个单细胞实验在广泛组织中的数据,包括数亿次测量。目前已经根据这些数据设计和训练了几个模型(例如scGPT、scFoundation、Geneformer),目的是为单细胞转录组数据创建一个基础模型,类似于自然语言处理中的基础模型。
提示微调
自GPT-2引入以来,提示已成为从大模型中引出有意义行为的常见方法。最近,公开可用的数据集,如Alpaca,和数据集生成器,如FLAN,以及参数有效的微调PEFT,使训练自定义大语言模型成为可能。
方法
数据转换
Cell2Sentence转换的核心是将细胞表达矩阵重组为降序排列转录物丰度的基因名称序列。令 C C C表示具有 n n n行 k k k个基因的计数矩阵, C i , j C_{i,j} Ci,j表示在细胞 i i i中观察到的基因 j j j的RNA分子的数量。作者遵循scRNA-seq数据的标准预处理步骤:过滤表达少于200个基因的细胞和过滤在少于200个细胞中表达的基因。然后使用Scanpy Python库计算质量控制指标,并过滤出含有2500个计数以上或线粒体基因转录物计数超过20%的低质量细胞。计数矩阵然后被row-标准化(每个细胞的转录求和为10,000),再被log-标准化,得到处理后的矩阵 C ′ C' C′,总结了标准化步骤为: C i , j ′ = l o g 10 ( 1 + 1 0 4 × C i , j ∑ j = 1 k C i , j ) C'_{i,j}=log_{10}(1+10^{4}\times \frac{C_{i,j}}{\sum_{j=1}^{k}C_{i,j}}) Ci,j′=log10(1+104×∑j=1kCi,jCi,j)然后将rank-order应用在 C ′ C' C′上得到 S S S,将 S ( C i ) S(C_{i}) S(Ci)产生的基因名称序列表示为细胞 i i i的细胞句子。在实践中,预处理和rank-order被应用在每个独立的数据集,为细胞句子生成提供了灵活性。
虽然基因在转录物基质中不是内在有序的,但它们的表达模式已被证明遵循rank频率模式(Furusawa&Kaneko,2003;邱等人,2013),因此,细胞内基因的表达水平与在该细胞中的基因rank之间建立了稳定的关系。作者用对数线性分布对这种rank关系进行建模,并使用线性回归在对数-对数空间中对其进行近似。
给定经过rank变换
S
S
S的单细胞数据集,设
r
i
r_i
ri表示基因
i
i
i在
C
′
C^′
C′中的rank的对数,
e
i
e_i
ei表示基因
i
i
i的原始表达。然后拟合线性模型,以在初始转换为细胞语句格式期间从
r
i
r_i
ri预测
e
i
e_i
ei,从而得到拟合的斜率和截距,并为每个转换的数据集保存(见图7)。对于给定的数据集
d
d
d,线性模型的参数
{
a
d
,
b
d
}
∈
R
2
\left\{a_{d},b_{d}\right\}\in\R^{2}
{ad,bd}∈R2和形式为:
e
i
=
a
d
×
r
i
+
b
i
e_{i}=a_{d}\times r_{i}+b_{i}
ei=ad×ri+bi

- 图7:rank与expression之间的关系。
通过忽略无效基因名称并平均重复基因的rank来对生成的细胞句子进行后处理。然后将拟合的线性模型应用于生成的基因的log-rank,以转换回表达。任何不存在于细胞句子中的基因都被认为是零表达的。作者将属于一组独特基因 G U ⊆ S G_U⊆S GU⊆S的生成基因 g i g e n g^{gen}_{i} gigen的平均rank定义如下: r i g e n = 1 ∣ G ∣ ∑ j = 1 ∣ G ∣ r a n k ( g j g e n ) r_{i}^{gen}=\frac{1}{|G|}\sum_{j=1}^{|G|}rank(g_{j}^{gen}) rigen=∣G∣1j=1∑∣G∣rank(gjgen)其中, G = { g 1 g e n , . . . , g n g e n } ⊆ S G=\left\{g_{1}^{gen},...,g_{n}^{gen}\right\}⊆S G={g1gen,...,gngen}⊆S是 g i g e n g_{i}^{gen} gigen的重复生成基因的集合。
因此,生成细胞的基因表达为:

在实践中,考虑单细胞数据集中所有基因名称的全局字典,它决定了细胞的最终基因表达载体的大小。由于线性模型只需要基因的log-rank来近似其表达水平,因此任何基因序列都可以转换为表达,包括那些生成的细胞句子。
任务
C2S包括3个任务:
- 生成细胞句子:C2S模型被训练为根据提示生成基因序列,可选地以额外的元数据为条件;
- 预测细胞标签:生物实验通常涉及组合标签(例如,患者和样本的元数据),C2S模型可以直接从文本中的细胞句子中学习预测这些标签;
- 衍生自然语言的见解:C2S分析单细胞数据,通过将相关自然语言与细胞句子配对,提取基因表达相关的人类可交互信息。
每个任务的提示和响应示例如图5所示。对于细胞生成和自然语言任务,使用标准因果语言建模损失来训练模型: L ( x ) = − 1 N ∑ i = 1 N l o g ( e x p ( z i v i ) ∑ v ∈ V e x p ( z i v ) ) L(x)=-\frac{1}{N}\sum_{i=1}^{N}log(\frac{exp(z_{i_{v_{i}}})}{\sum_{v\in V}exp(z_{i_{v}})}) L(x)=−N1i=1∑Nlog(∑v∈Vexp(ziv)exp(zivi))其中 x = { x 0 , . . . , x N } x=\left\{x_{0},...,x_{N}\right\} x={x0,...,xN}是input sentence( N + 1 N+1 N+1个tokens),模型在位置 i i i输出 z i ∈ R ∣ V ∣ z_{i}\in\R^{|V|} zi∈R∣V∣, V V V是语言模型的词汇表, v i v_i vi是 x x x的标记化中位置 i i i处的GT token。损失在每个批次上取平均值。

- 图5:Cell2Sentence的提示和响应示例,用于从文本生成细胞句子、预测复杂的自然语言标签以及从单个细胞句子生成生物学见解。
实验
实验是使用免疫组织数据集(Dom´ınguez-Conde-2022)或大规模多组织数据集(Megill-2021)进行微调的模型进行的。在这两种情况下,由于资源限制,使用截断为100个基因的细胞句子来微调GPT-2(小型、中型和大型)。还微调了Pythia-16m,它基于GPT NeoX架构,并使用旋转嵌入(rotary embeddings)。对于后者,在微调期间将模型的最大输入序列长度设置为9200个token,允许操作full cell sentence。首先在大型单细胞数据集上微调语言模型,然后再可选地在指定数据集继续训练以评估。
微调数据集
作者将实验重点放在三个具有广泛自然语言元数据和标签的数据集上,从而能够利用基本模型的功能。
Immune tissue
免疫组织(Dom´ınguez-Conde-2022)提出了一个具有细胞类型注释的大型人类免疫组织单细胞数据集。数据转换后,可以获得273,502个细胞句子,每个句子与35个细胞类型标签中的一个配对。作者保留了20%的细胞句子用于验证(10%)和测试(10%)。该数据集有三个任务:
- 无条件细胞生成(生成一个没有任何指定细胞标签的随机细胞)
- 细胞类型生成(生成给定特定细胞类型的细胞句子)
- 细胞类型预测(在给定细胞句子提示的情况下,用自然语言预测细胞类型)
作者创建了20个提示模板,用于在自然语言中嵌入细胞句子和标签。
Cytokine stimulation
细胞因子刺激(Dong-2023)是一个单细胞数据集,将9种细胞因子刺激组合应用于免疫组织,并有2种不同的exposure。将细胞分成7种细胞类型,总共140个组合labels,包括未刺激的对照细胞。此数据集用于两个任务:
- 扰动细胞生成(仅在给定文本格式标签的情况下生成扰动细胞)
- 细胞标签分类(根据输入细胞对细胞类型、扰动和exposure进行分类)
在扰动细胞生成任务中,140个组合标签中的10个在训练期间被保留,以便在测试时使用。对于细胞标签分类,所有组合都在训练期间使用,但在训练期间只使用了有限的数据量。
Multi-tissue
多组织(Megill-2021)提供了对数百个人类和小鼠单细胞数据集的访问。作者选择了99个人类单细胞数据集,并将每个数据集转换为细胞句子,共产生3700万个细胞(包括19项已完成的研究,占所有细胞句子的2.7%)。每个细胞句子都与来自研究元数据的组织标签配对(例如,包含“大脑”和“肝脏”细胞的数据集的组织标签将为“大脑,肝脏”)。作者在这个数据集中总共发现了11种独特的组织和42种独特的结构组合。作者为这个多组织数据集推导出以下任务:
- 组织类型预测和条件生成(类似于之前的细胞类型预测和有条件生成提示)
- 从细胞句子生成摘要(由细胞句子提示,模型为相应的研究生成摘要)
- 生成一个给定摘要的细胞句子(类似于从组织或细胞类型生成,但这里利用摘要中的自然语言)
训练的相关内容
作者将自然语言称为“NL”,将Cell2Sentence称为“C2S”,并将仅在细胞句子上训练的模型称为“C2S”,与称之为“NL+C2S”的在细胞句子中微调的预训练模型形成对比。作者对GPT-2使用1024的序列长度,对Pythia-16m使用9200的序列长度并在所有token上进行训练。作者使用AdamW优化器和flash attention。作者发现,C2S模型在很大程度上受益于自然语言预训练,而不是从随机初始化的权重开始训练(由于C2S其实不要求自然语言上预训练,所以模型embedding size可以设置小一点,从而减少参数)。
方式1:C2S预训练
GPT-2小模型初始化为12层和768个hidden维度,中模型初始化为24层和1024个hidden维度。
作者在full cell sentence数据集上训练字节对编码(BPE)标记器,包括NL提示和细胞类型标签,产生9609个token的词汇表。训练集包含大约3000万个tokens,平均每个example有740个tokens。由于可以选择较小的嵌入空间,初始化的模型包含的参数比在50257个tokens的词汇表上预训练的对应模型略少(小模型93M,中等模型313M)。
损失是根据提示和相关标签(即细胞类型)计算的。不这样做将会削弱模型学习提示token和标签token之间的条件关系的能力。
方式2:NL+C2S微调
使用Hugging Face的预训练权重对模型进行初始化。我们采用余弦调度器。作者尝试过应用有效的微调技术(例如LoRA),完全微调的模型优于LoRA微调。值得注意的是,LoRA产生了高度可变的生成模式,生成句子中基因的唯一性低至70%。与预训练设置不同,这里以经典的方式应用指令微调任务,只计算标签上的损失。
实验1:条件细胞生成
细胞类型生成
在来自(Dom´ınguez-Conde-2022)的免疫组织数据集上训练C2S。细胞句子被附加到指示其细胞类型的文本提示中,以便模型学习细胞类型生成(图5)。C2S与几种已建立的生成单细胞方法进行了比较,包括scVI、scGen、scDiffusion和scGPT。
扰动细胞生成
使用来自(Dong-2023)的细胞因子刺激数据集。提示的构建类似于细胞类型生成任务,除了具有额外的细胞因子刺激和exposure标签,每个细胞句子总共有3个标签。使用scanpy对标准过滤后剩余的21710个基因进行C2S和scGen模型的训练。传统的扰动细胞生成是:
- 给定对照样本的基因表达,以元数据为条件,生成扰动后的基因表达
- 在C2S中,由于没有基因表达,而是细胞句子,所以选择了一个粗略的表示方法,即根据元数据生成细胞句子
- 或许有更合适的改进方法:给定对照细胞的细胞句子,结合元数据,生成新的细胞句子
后续实验其实与之类似,关键在于模板的获取,比如对于摘要生成,模板就应该是摘要+细胞句子。










![[汇总] Docker容器详解 Macvlan 创建不同容器独立跑仿真(持续更新中)](https://img-blog.csdnimg.cn/direct/3aa76f80bc134f838599f2ae0ec3bb1b.png)

