基于yaml或ini配置文件,配置文件包含例如样本名称、参考基因组版本、exon capture bed文件路径、参考基因组路径和ANNOVAR注释文件等信息。
基于该流程可以实现全外显测序的fastq文件输入到得到最终变异VCF文件。
1. Snakemake分析流程基础软件安装
# conda安装
conda install -c bioconda snakemake -y
conda install -c bioconda fastqc -y
conda install -c bioconda trim-galore -y
conda install -c bioconda bwa -y
conda install -c bioconda samtools -y
# GATK 4.2.1版本
conda install -c bioconda gatk -y
ANNOVAR注释软件安装和使用参考文章:
https://www.jianshu.com/p/461f2cd47564
ANNOVAR注释全外显子有些坑和经常会报错的问题,
本人后续有空会写下对全外显子注释的ANNOVAR软件安装和使用:
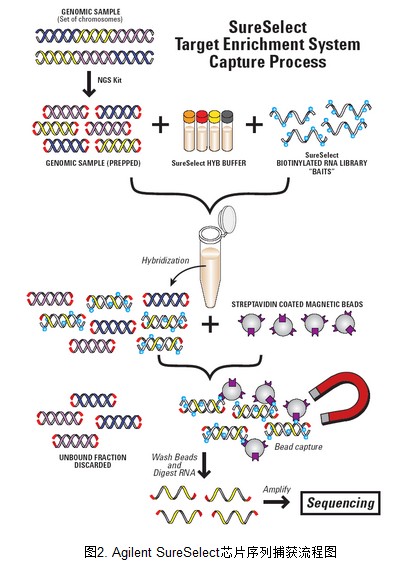
2. AgilentSureSelect 外显子序列捕获流程图
外显子捕获基本原理参考下图。
3. GATK SNP和INDEL分析基本流程
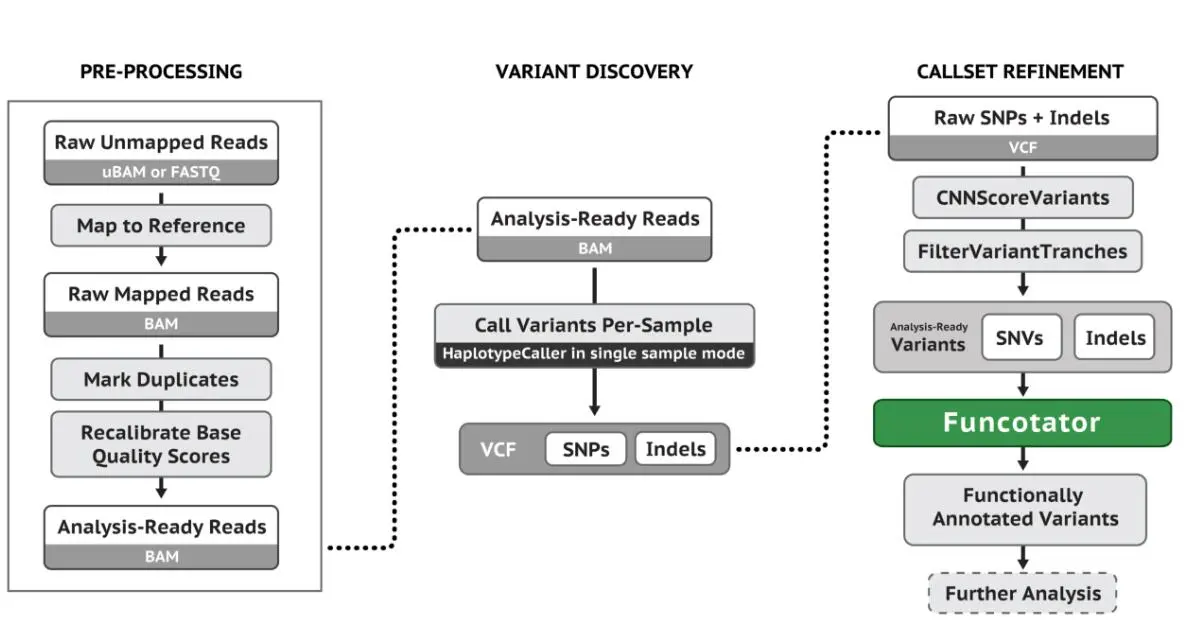
4. Snakemake全外显子分析流程
其中regions_bed配置为外显子捕获所采用试剂盒对应的bed文件路径。
##################### parser configfile #########################
import os
configfile: "config.yaml"
# configfile: "config.ini"
sample = config["sample"]
genome = config["genome"]
#################################################################
PROJECT_DATA_DIR = config["project_data_dir"]
PROJECT_RUN_DIR = config["project_run_dir"]
os.system("mkdir -p {0}".format(PROJECT_RUN_DIR))
######################### Output #################################
ALL_FASTQC_RAW_ZIP1 = PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_1_fastqc.zip"
ALL_FASTQC_RAW_HTML1 = PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_1_fastqc.html"
ALL_FASTQC_RAW_ZIP2 = PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_2_fastqc.zip"
ALL_FASTQC_RAW_HTML2 = PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_2_fastqc.html"
ALL_CELAN_FASTQ1 = PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_1_val_1.fq.gz"
ALL_CELAN_FASTQ2 = PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_2_val_2.fq.gz"
ALL_SORTED_BAM = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam"
ALL_SORTED_BAM_BAI1 = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam.bai"
ALL_BAM_FLAGSTAT = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam.flagstat"
ALL_EXON_INTERVAL_BED = PROJECT_RUN_DIR + "/gatk/" + sample + ".Exon.Interval.bed"
ALL_STAT_TXT = PROJECT_RUN_DIR + "/gatk/" + sample + ".stat.txt"
ALL_MKDUP_BAM = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam"
ALL_METRICS = PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.bam.metrics"
ALL_SORTED_BAM_BAI2 = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam.bai"
ALL_INSERT_SIZE_HISTOGRAM_pdf = PROJECT_RUN_DIR + "/gatk/" + sample + ".insert_size_histogram.pdf"
ALL_RECAL_DATA_TABLE = PROJECT_RUN_DIR + "/gatk/" + sample + ".recal_data.table"
ALL_BQSR_SORTED_BAM = PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.mkdup.BQSR.bam"
ALL_depthOfcoverage = PROJECT_RUN_DIR + "/gatk/depthOfcoverage"
ALL_RAW_VCF = PROJECT_RUN_DIR + "/vcf/" + sample + ".raw.vcf"
All_SNP_SELECT = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.vcf"
ALL_SNP_FILTER_VCF = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.vcf"
ALL_SNP_FILTER_VCF_STAT = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.filter.vcf_stat.csv"
All_INDEL_SELECT = PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.vcf"
ALL_INDEL_FILTER_VCF = PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.vcf"
ALL_SNP_AVINPUT = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.avinput"
ALL_INDEL_AVINPUT = PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.avinput"
ALL_MERG_FILTERE_VCF = PROJECT_RUN_DIR + "/vcf/" + sample + ".merge.filter.vcf"
ALL_SNPANNO = PROJECT_RUN_DIR + "/vcf/" + sample + "_snpanno." + genome + "_multianno.csv"
ALL_INDELANNO = PROJECT_RUN_DIR + "/vcf/" + sample + "_indelanno." + genome + "_multianno.csv"
ALL_ALLANNO = PROJECT_RUN_DIR + "/vcf/" + sample + "_allanno." + genome + "_multianno.csv"
QUALIMAP_REPORT = PROJECT_RUN_DIR + "/gatk/bam_QC/" + sample + "_bamQC_report.pdf"
######################## Workflow #################################
rule all:
input:
ALL_FASTQC_RAW_ZIP1,
ALL_FASTQC_RAW_HTML1,
ALL_FASTQC_RAW_ZIP2,
ALL_FASTQC_RAW_HTML2,
ALL_SORTED_BAM,
ALL_SORTED_BAM_BAI1,
ALL_BAM_FLAGSTAT,
ALL_EXON_INTERVAL_BED,
ALL_STAT_TXT,
ALL_MKDUP_BAM,
ALL_METRICS,
ALL_SORTED_BAM_BAI2,
ALL_RECAL_DATA_TABLE,
ALL_BQSR_SORTED_BAM,
ALL_RAW_VCF,
All_SNP_SELECT,
All_INDEL_SELECT,
ALL_SNP_FILTER_VCF,
ALL_INDEL_FILTER_VCF,
ALL_MERG_FILTERE_VCF,
ALL_SNPANNO,
ALL_INDELANNO,
ALL_ALLANNO
rule fastqc_raw:
input:
r1 = PROJECT_DATA_DIR + "/" + sample + "_1.fq.gz",
r2 = PROJECT_DATA_DIR + "/" + sample + "_2.fq.gz"
output:
PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_1_fastqc.zip",
PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_2_fastqc.zip",
PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_1_fastqc.html",
PROJECT_RUN_DIR + "/fastqc/raw_data/" + sample + "_2_fastqc.html"
threads: 8
params:
raw_data_dir = PROJECT_RUN_DIR + "/fastqc/raw_data",
clean_data_dir = PROJECT_RUN_DIR + "/fastqc/clean_data",
align_dir = PROJECT_RUN_DIR + "/align",
gatk_dir = PROJECT_RUN_DIR + "/gatk",
vcf_dir = PROJECT_RUN_DIR + "/vcf",
tmp_dir = PROJECT_RUN_DIR + "/tmp",
fastqc_report_dir = PROJECT_RUN_DIR + "/report/fastqc_report",
align_report_dir = PROJECT_RUN_DIR + "/report/align_report"
shell:
"""
mkdir -p {params.raw_data_dir}
mkdir -p {params.clean_data_dir}
mkdir -p {params.align_dir}
mkdir -p {params.gatk_dir}
mkdir -p {params.vcf_dir}
mkdir -p {params.tmp_dir}
fastqc -f fastq --extract -t {threads} -o {params.raw_data_dir} {input.r1} {input.r2}
"""
rule fastqc_clean:
input:
r1 = PROJECT_DATA_DIR + "/" + sample + "_1.fq.gz",
r2 = PROJECT_DATA_DIR + "/" + sample + "_2.fq.gz"
output:
temp(PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_1_val_1.fq.gz"),
temp(PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_2_val_2.fq.gz")
threads: 8
params:
quality = 20,
length = 20,
clean_data_dir = PROJECT_RUN_DIR + "/fastqc/clean_data"
shell:
"""
trim_galore --paired --quality {params.quality} --length {params.length} -o {params.clean_data_dir} {input.r1} {input.r2}
"""
# BWA mem比对
rule bwa_mem:
input:
reads1 = PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_1_val_1.fq.gz",
reads2 = PROJECT_RUN_DIR + "/fastqc/clean_data/" + sample + "_2_val_2.fq.gz"
output:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam"
threads:8
params:
hg = config["hg_fa"]
shell:
"""
bwa mem -t {threads} -M -R "@RG\\tID:{sample}\\tPL:ILLUMINA\\tLB:{sample}\\tSM:{sample}" {params.hg} {input.reads1} {input.reads2}|samtools sort -@ {threads} -o {output}
"""
rule bam_index_1:
input:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam"
output:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam.bai"
threads: 2
shell:
"samtools index {input} > {output}"
rule flagstat:
input:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam"
output:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam.flagstat"
threads: 1
shell:
"samtools flagstat {input} > {output}"
# 创建reference fasta 字典,GATK需要用到
# rule CreateSequenceDictionary:
# input:
# "/reference/hg19/ucsc.hg19.fa"
# output:
# PROJECT_RUN_DIR + "/" + sample + "/gatk/" + sample + ".hg19.dict"
# # /GATK_bundle/hg19/ucsc.hg19.dict
# threads: 1
# params:
# gatk4 = "/public/software/gatk-4.0.6.0/gatk",
# gatk_dir = PROJECT_RUN_DIR + "/" + sample + "/gatk"
# shell:
# """
# mkdir -p {params.gatk_dir}
# gatk CreateSequenceDictionary -R {input} -O {ouptut}
# """
rule CreateExonIntervalBed:
input:
regions_bed = config["regions_bed"],
hg_dict = config["hg_dict"]
output:
Exon_Interval_bed = PROJECT_RUN_DIR + "/gatk/" + sample + ".Exon.Interval.bed"
threads: 1
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk"
shell:
"""
gatk BedToIntervalList -I {input.regions_bed} -O {output.Exon_Interval_bed} -SD {input.hg_dict}
"""
# 外显子区域覆盖度
rule exon_region:
input:
bam = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam",
Exon_Interval_bed = PROJECT_RUN_DIR + "/gatk/" + sample + ".Exon.Interval.bed"
output:
stat_txt = PROJECT_RUN_DIR + "/gatk/" + sample + ".stat.txt"
threads: 2
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk"
shell:
"""
gatk CollectHsMetrics -BI {input.Exon_Interval_bed} -TI {input.Exon_Interval_bed} -I {input.bam} -O {output.stat_txt}
"""
# 标记PCR重复序列
rule mark_duplicates:
input:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.bam"
output:
mkdup_bam = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam",
metrics = PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.bam.metrics"
threads: 4
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk"
shell:
"""
gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" MarkDuplicates -I {input} -O {output.mkdup_bam} -M {output.metrics}
"""
rule bam_index_2:
input:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam"
output:
PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam.bai"
threads: 2
shell:
"samtools index {input} > {output}"
# 插入片段分布统计
#rule InsertSizeStat:
# input:
# PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.mkdup.bam"
# output:
# insert_size_metrics_txt = temp(PROJECT_RUN_DIR + "/gatk/" + sample + ".insert_size_metrics.txt"),
# insert_size_histogram_pdf = PROJECT_RUN_DIR + "/gatk/" + sample + ".insert_size_histogram.pdf"
# threads:1
# params:
# picard = "/public/software/picard.jar",
# M = 0.5
# shell:
# """
# java -jar {params.picard} CollectInsertSizeMetrics \
# I={input} \
# O={output.insert_size_metrics_txt} \
# H={output.insert_size_histogram_pdf} \
# M=0.5
# """
# 重新校正碱基质量值
# 计算出所有需要进行重校正的read和特征值,输出为一份校准表文件.recal_data.table
rule BaseRecalibrator:
input:
mkdup_bam = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam",
mkdup_bam_index = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam.bai",
vcf_1000G_phase1_indels_sites = config["vcf_1000G_phase1_indels_sites"],
vcf_Mills_and_1000G_gold_standard_indels_sites = config["vcf_Mills_and_1000G_gold_standard_indels_sites"],
vcf_dbsnp = config["vcf_dbsnp"]
output:
PROJECT_RUN_DIR + "/gatk/" + sample + ".recal_data.table"
threads: 8
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk",
hg_fa = config["hg_fa"],
regions_bed = config["regions_bed"]
shell:
"""
gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" BaseRecalibrator \
-R {params.hg_fa} \
-I {input.mkdup_bam} \
--known-sites {input.vcf_1000G_phase1_indels_sites} \
--known-sites {input.vcf_Mills_and_1000G_gold_standard_indels_sites} \
--known-sites {input.vcf_dbsnp} \
-L {params.regions_bed} \
-O {output}
"""
# gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" BaseRecalibrator -R genome/hg19.fa -I ./TKQX.sorted.mkdup.bam --known-sites /GATK_bundle/hg19/1000G_phase1.indels.hg19.sites.vcf --known-sites /GATK_bundle/hg19/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf --known-sites /GATK_bundle/hg19/dbsnp_138.hg19.vcf -L /GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed -O ./TKQX.recal_data.table
# java -jar CreateSequenceDictionary.jar R= Homo_sapiens_assembly18.fasta O= Homo_sapiens_assembly18.dict
# 利用校准表文件重新调整原来BAM文件中的碱基质量值,使用这个新的质量值重新输出新的BAM文件
rule ApplyBQSR:
input:
mkdup_bam = PROJECT_RUN_DIR + "/align/" + sample + ".sorted.mkdup.bam",
recal_data = PROJECT_RUN_DIR + "/gatk/" + sample + ".recal_data.table",
regions_bed = config["regions_bed"]
#regions_bed = "/GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed"
output:
PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.mkdup.BQSR.bam"
threads: 4
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk",
hg_fa = config["hg_fa"]
#hg_fa = "genome/hg19.fa"
shell:
"""
gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" ApplyBQSR \
-R {params.hg_fa} \
-I {input.mkdup_bam} \
-bqsr {input.recal_data} \
-L {input.regions_bed} \
-O {output}
"""
# gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" ApplyBQSR -R genome/hg19.fa -I ./test_sample.sorted.mkdup.bam -bqsr ./test_sample.recal_data.table -L /GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed -O ./test_sample.sorted.mkdup.BQSR.bam
# # 校正碱基图
# rule recal_data_plot:
# input:
# PROJECT_RUN_DIR + "/" + sample + "/gatk/" + sample + ".recal_data.table"
# output:
# PROJECT_RUN_DIR + "/" + sample + "/gatk/" + sample + ".recal_data.table.plot"
# threads: 1
# params:
# gatk4 = "/public/software/gatk-4.0.6.0/gatk",
# gatk = "/public/software/anaconda3/envs/PGS_1M/bin/gatk"
# shell:
# """
# gatk AnalyzeCovariates -bqsr {input} -plots {output}
# """
# gatk AnalyzeCovariates -bqsr ./test_sample.recal_data.table -plots ./test_sample.recal_data.table.plot
# 统计测序深度和覆盖度
#rule depthOfcoverage:
# input:
# BQSR_mkdup_bam = PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.mkdup.BQSR.bam"
# output:
# PROJECT_RUN_DIR + "/gatk/depthOfcoverage"
# threads:1
# params:
# #hg19_fa = "genome/hg19.fa",
# hg_fa = config["hg_fa"],
# bamdst = "/public/software/bamdst/bamdst",
# regions_bed = config["regions_bed"]
# #regions_bed = "/GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed"
# shell:
# """
# {params.bamdst} -p {params.regions_bed} -o {output} {input.BQSR_mkdup_bam}
# """
# """
# gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" DepthOfCoverage \
# -R {params.hg19} \
# -o {output} \
# -I {input.BQSR_mkdup_bam} \
# -L {input.regions_bed} \
# --omitDepthOutputAtEachBase --omitIntervalStatistics \
# -ct 1 -ct 5 -ct 10 -ct 20 -ct 30 -ct 50 -ct 100
# """
#gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" DepthOfCoverage -R genome/hg19.fa -o ./TKQX -I ./TKQX.sorted.mkdup.BQSR.bam -L /GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 5 -ct 10 -ct 20 -ct 30 -ct 50 -ct 100
# /public/software/bamdst/bamdst -p /GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed -o ./depth_coverage ./TKQX.sorted.mkdup.BQSR.bam
# # 单样本VCF calling
rule vcf_calling:
input:
BQSR_mkdup_bam = PROJECT_RUN_DIR + "/gatk/" + sample + ".sorted.mkdup.BQSR.bam"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".raw.vcf"
threads: 4
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk",
hg_fa = config["hg_fa"],
vcf_dbsnp = config["vcf_dbsnp"],
regions_bed = config["regions_bed"]
shell:
"""
gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" HaplotypeCaller \
-R {params.hg_fa} \
-I {input.BQSR_mkdup_bam} \
-D {params.vcf_dbsnp} \
-L {params.regions_bed} \
-O {output}
"""
# gatk --java-options "-Xmx30G -Djava.io.tmpdir=./" HaplotypeCaller -R genome/hg19.fa -I ./TKQX.sorted.mkdup.BQSR.bam -D /GATK_bundle/hg19/dbsnp_138.hg19.vcf -L /GATK_bundle/bed/SureSelectAllExonV6/S07604514_Regions.bed -O ./TKQX.raw.vcf
# 变异质控和过滤,对raw data进行质控,剔除假阳性的标记
rule vcf_select_SNP:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".raw.vcf"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.vcf"
threads: 2
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk"
shell:
"""
gatk SelectVariants -select-type SNP -V {input} -O {output}
"""
rule vcf_filter_SNP:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.vcf"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.filter.vcf"
threads: 2
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk",
QD = 2.0,
MQ = 40.0,
FS = 60.0,
SOR = 3.0,
MQRankSum = -12.5,
ReadPosRankSum = -8.0
shell:
"""
gatk VariantFiltration \
-V {input} \
--filter-expression "QD < {params.QD} || MQ < {params.MQ} || FS > {params.FS} || SOR > {params.SOR} || MQRankSum < {params.MQRankSum} || ReadPosRankSum < {params.ReadPosRankSum}" --filter-name "PASS" \
-O {output}
"""
# --filter-expression "QD < 2.0|| MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" --filter-name "PASS"‘
# gatk VariantFiltration -V TKQX.snp.vcf --filter-expression "QD < 2.0|| MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" --filter-name "PASS" -O TKQX.snp.filter.vcf
# rule snp_frequency_stat:
# input:
# PROJECT_RUN_DIR + "/" + sample + "/vcf/" + sample + ".snp.filter.vcf"
# output:
# PROJECT_RUN_DIR + "/" + sample + "/vcf/" + sample + ".snp.filter.vcf_stat.csv"
# threads: 1
# params:
# gatk4 = "/public/software/gatk-4.0.6.0/gatk",
# snp_frequency = "/public/analysis/pipeline/WES/scripts/snp_frequency.py"
# shell:
# """
# python {params.snp_frequency} {input}
# """
rule vcf_select_InDel:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".raw.vcf"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.vcf"
threads: 2
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk"
shell:
"""
gatk SelectVariants -select-type INDEL -V {input} -O {output}
"""
rule vcf_filter_InDel:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.vcf"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.filter.vcf"
threads: 2
params:
gatk4 = "/public/software/gatk-4.0.6.0/gatk",
QD = 2.0,
FS = 200.0,
SOR = 10.0,
MQRankSum = -12.5,
ReadPosRankSum = -8.0
shell:
"""
gatk VariantFiltration \
-V {input} \
--filter-expression "QD < {params.QD} || FS > {params.FS} || SOR > {params.SOR} || MQRankSum < {params.MQRankSum} || ReadPosRankSum < {params.ReadPosRankSum}" --filter-name "PASS" \
-O {output}
"""
# --filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0|| MQRankSum < -12.5 || ReadPosRankSum < -8.0" --filter-name "PASS"
rule merge_vcf:
input:
snp_filter_vcf = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.filter.vcf",
indel_filter_vcf = PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.filter.vcf"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + ".merge.filter.vcf"
threads: 1
params:
vcf_dir = PROJECT_RUN_DIR + "/vcf"
shell:
"""
cd {params.vcf_dir}
gatk MergeVcfs \
-I {input.snp_filter_vcf} \
-I {input.indel_filter_vcf} \
-O {output}
"""
rule transfer_avinput:
input:
snp = PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.filter.vcf",
indel= PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.filter.vcf",
merge = PROJECT_RUN_DIR + "/vcf/" + sample + ".merge.filter.vcf"
output:
snp = temp(PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.avinput"),
indel = temp(PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.avinput"),
merge = temp(PROJECT_RUN_DIR + "/vcf/" + sample + ".merge.avinput")
threads: 4
params:
convert2annovar = "/public/software/annovar/convert2annovar.pl"
shell:
"""
perl {params.convert2annovar} -format vcf4 {input.snp} > {output.snp}
perl {params.convert2annovar} -format vcf4 {input.indel} > {output.indel}
perl {params.convert2annovar} -format vcf4 {input.merge} > {output.merge}
"""
# SNP注释
rule snp_annotate:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".snp.avinput"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + "_snpanno." + genome + "_multianno.csv"
threads: 2
params:
table_annovar = "/public/software/annovar/table_annovar.pl",
humandb = "/public/software/annovar/humandb/",
prefix = sample + "_snpanno",
genome = config["genome"],
vcf_dir = PROJECT_RUN_DIR + "/vcf"
shell:
"""
cd {params.vcf_dir}
perl {params.table_annovar} {input} {params.humandb} -buildver {params.genome} -out {params.prefix} \
-remove -protocol refGene,cytoBand,clinvar_20221231,avsnp147,exac03,gnomad211_exome,SAS.sites.2015_08,esp6500siv2_all,intervar_20180118,dbnsfp42c \
-operation g,r,f,f,f,f,f,f,f,f -nastring . -csvout
"""
# INDEL注释
rule indel_annotate:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".indel.avinput"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + "_indelanno." + genome + "_multianno.csv"
threads: 2
params:
table_annovar = "/public/software/annovar/table_annovar.pl",
humandb = "/public/software/annovar/humandb/",
prefix = sample + "_indelanno",
genome = config["genome"],
vcf_dir = PROJECT_RUN_DIR + "/vcf",
perl = "/home/miniconda3/pkgs/perl-5.26.2-h36c2ea0_1008/bin/perl"
shell:
"""
cd {params.vcf_dir}
perl {params.table_annovar} {input} {params.humandb} -buildver {params.genome} -out {params.prefix} \
-remove -protocol refGene,cytoBand,clinvar_20221231,gnomad211_exome,exac03,esp6500siv2_all,ALL.sites.2015_08 \
-operation g,r,f,f,f,f,f -nastring . -csvout
"""
#
rule merge_annotate:
input:
PROJECT_RUN_DIR + "/vcf/" + sample + ".merge.avinput"
output:
PROJECT_RUN_DIR + "/vcf/" + sample + "_allanno." + genome + "_multianno.csv"
threads: 2
params:
table_annovar = "/public/software/annovar/table_annovar.pl",
humandb = "/public/software/annovar/humandb/",
prefix = sample + "_allanno",
genome = config["genome"],
vcf_dir = PROJECT_RUN_DIR + "/vcf"
shell:
"""
cd {params.vcf_dir}
perl {params.table_annovar} {input} {params.humandb} -buildver {params.genome} -out {params.prefix} \
-remove -protocol refGene,cytoBand,clinvar_20221231,avsnp147,exac03,gnomad211_exome,SAS.sites.2015_08,esp6500siv2_all,intervar_20180118,dbnsfp42c \
-operation g,r,f,f,f,f,f,f,f,f -nastring . -csvout
"""
生信分析进阶文章推荐
生信分析进阶1 - HLA分析的HLA区域reads提取及bam转换fastq
生信分析进阶2 - 利用GC含量的Loess回归矫正reads数量
生信分析进阶3 - pysam操作bam文件统计unique reads和mapped reads高级技巧合辑
生信分析进阶4 - 比对结果的FLAG和CIGAR信息含义与BAM文件指定区域提取