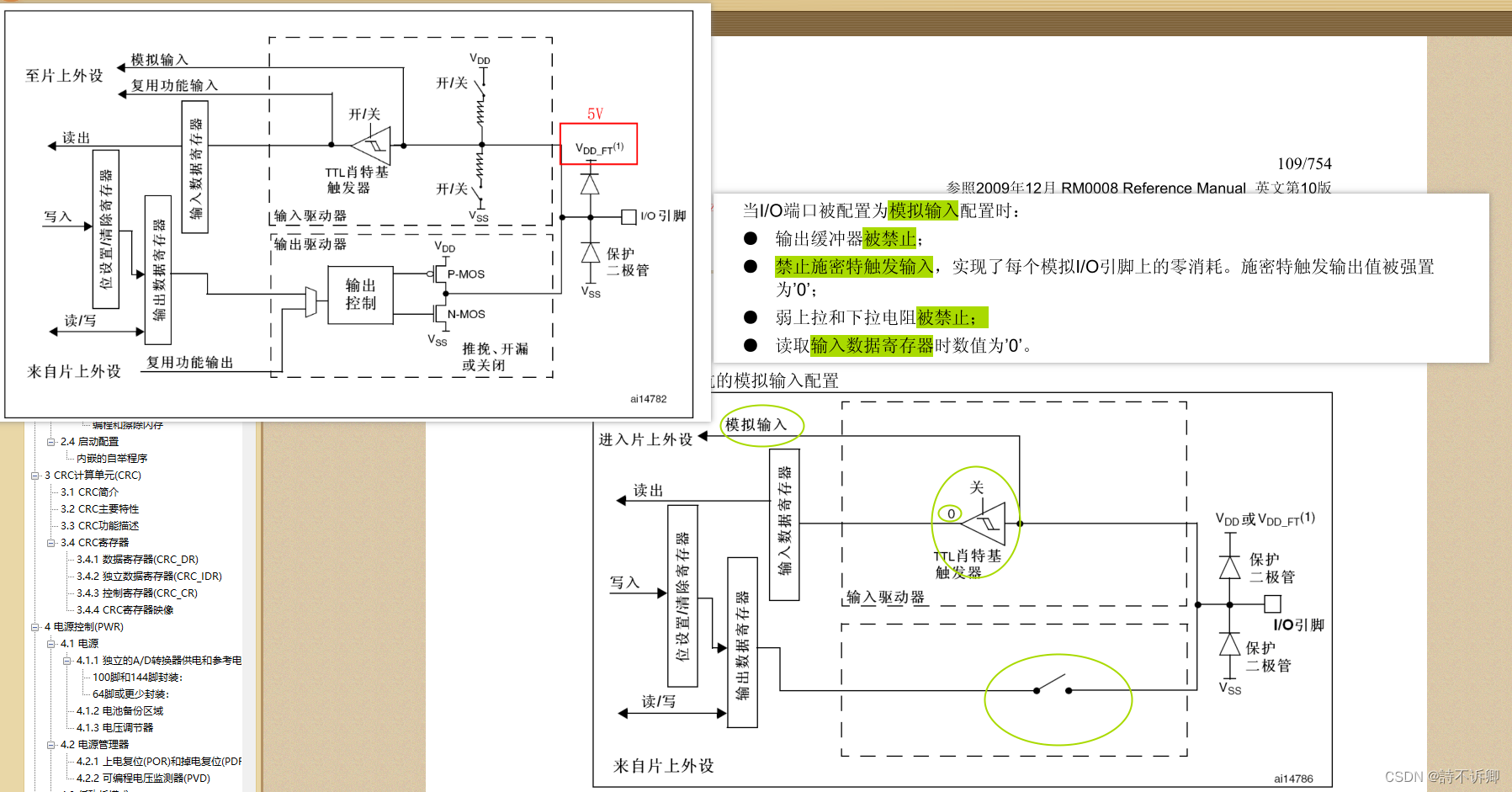
参考:https://www.bilibili.com/video/BV1sq4y1C7Qx/
https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html
代码下载:scanpy分析scRNA-seq数据基本流程(含scanpy seurat两大工具对比) 链接: https://pan.baidu.com/s/1vImSP_MNKEHuef-1qXE0pQ?pwd=9yvf 提取码: 9yvf
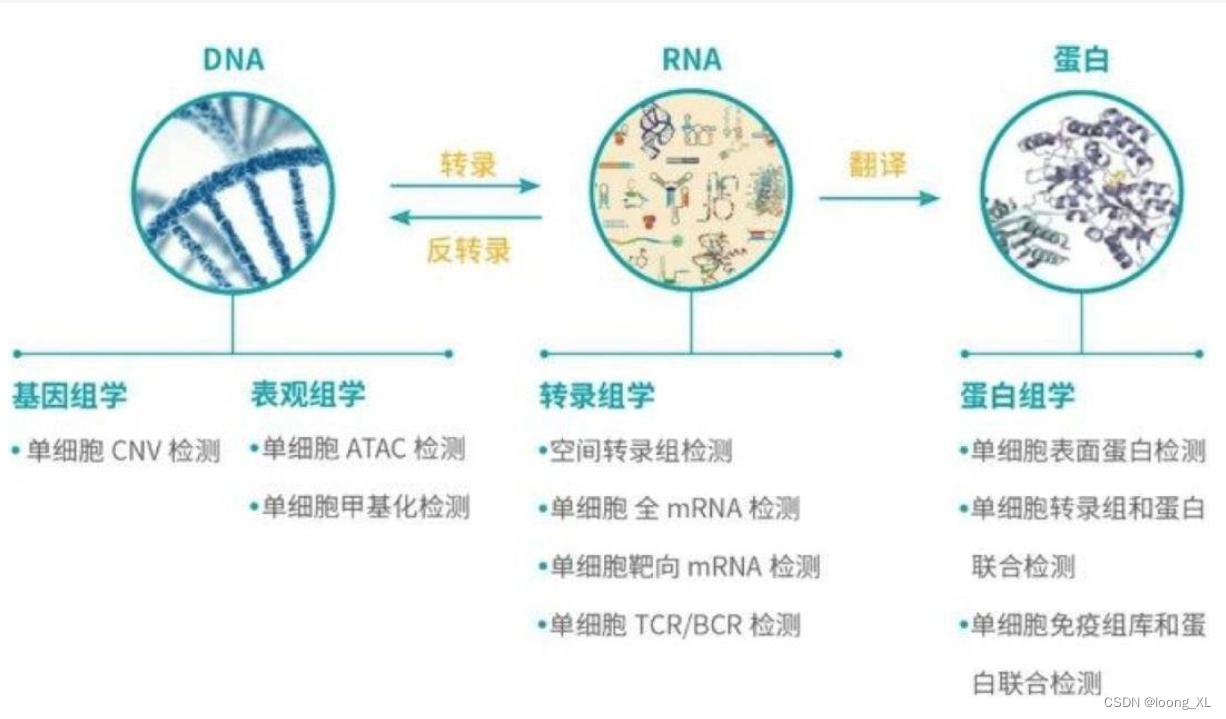
一般分析流程
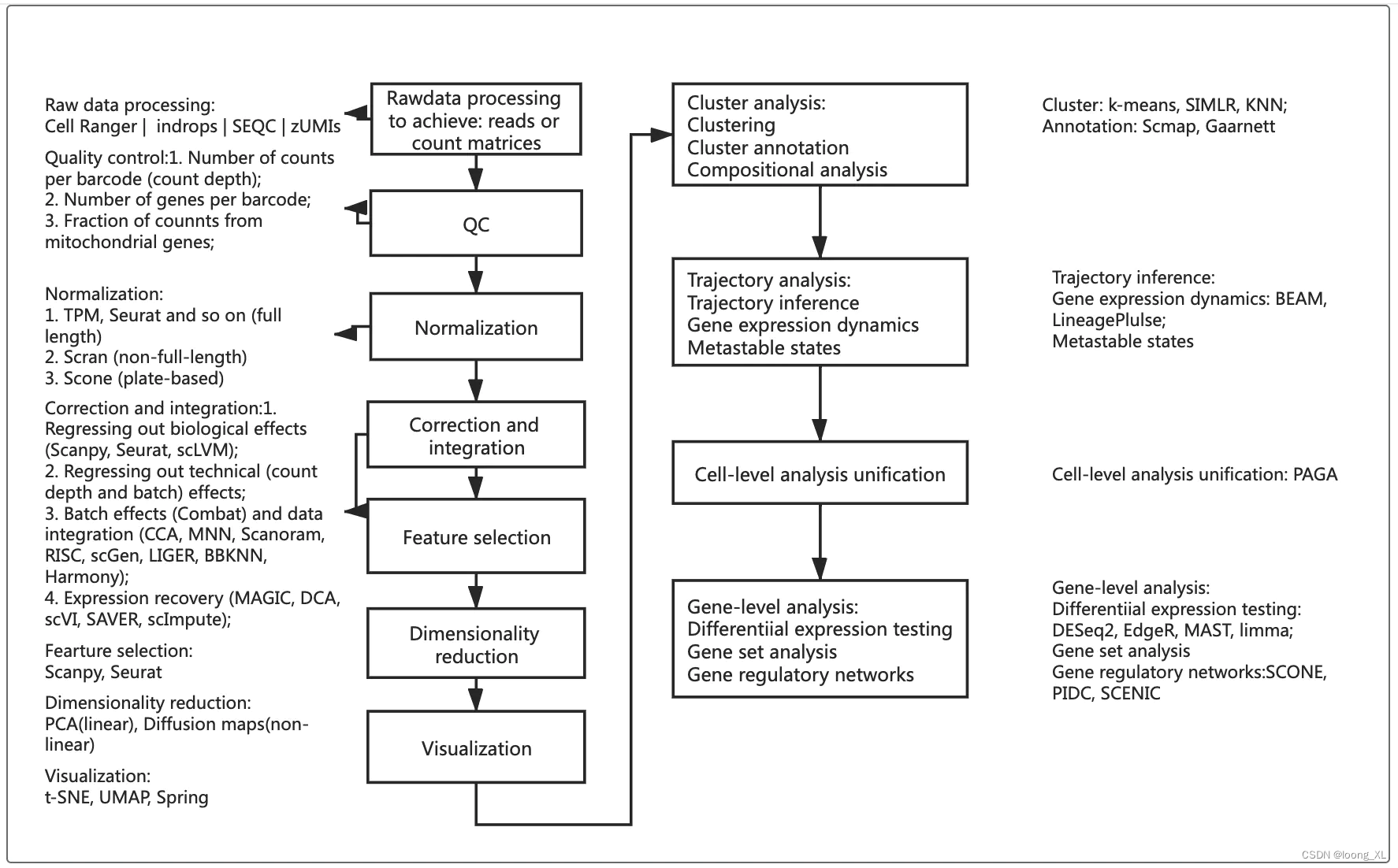
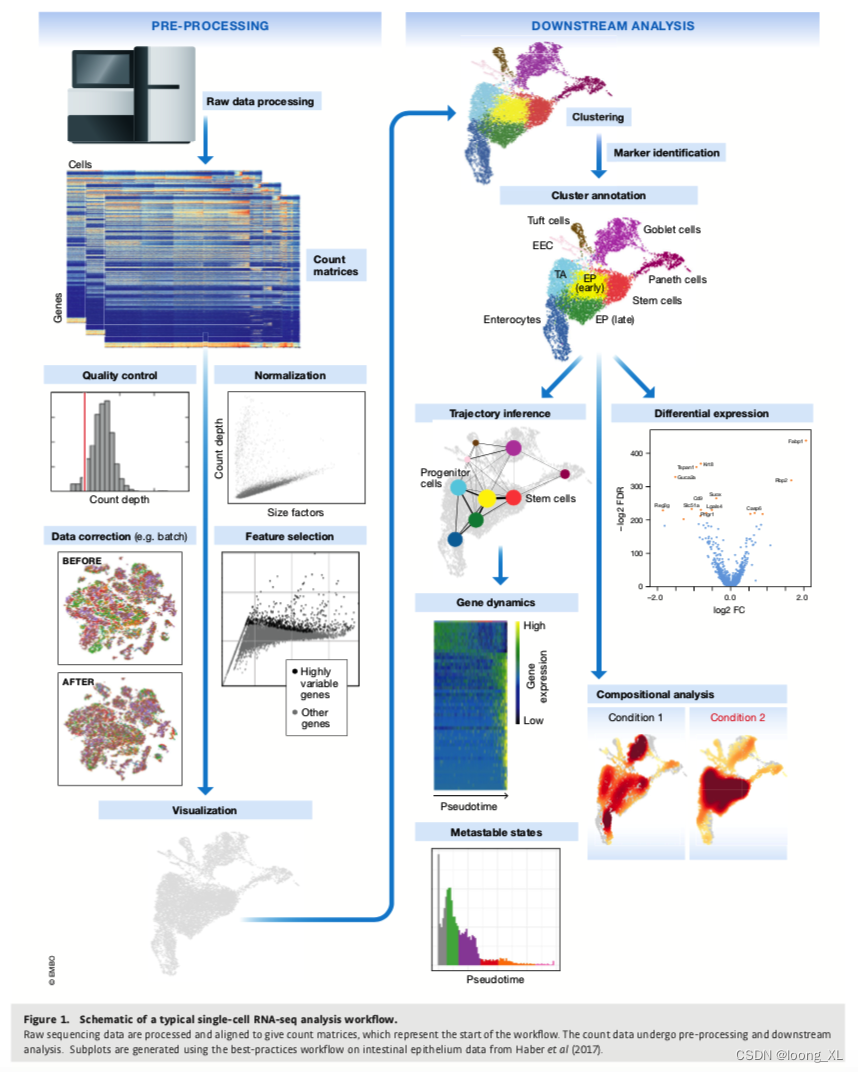
简单的示例,它读入了单细胞数据,进行了预处理,降维和聚类,最后可视化
1、读取数据: 使用Scanpy的read_10x_mtx函数读取单细胞数据。这里的输入数据是一个10X格式的矩阵文件,它包含了基因表达数据。2、数据预处理:
使用Scanpy的filter_genes函数过滤掉在少于3个细胞中表达量为0的基因,使用normalize_per_cell函数对每个细胞的基因表达数据进行标准化,使用log1p函数对基因表达数据进行log转换。3、降维: 使用Scanpy的tl.pca函数对数据进行降维,将高维的基因表达数据映射到二维或三维空间中。
4、聚类: 使用Scanpy的tl.louvain函数对数据进行聚类。
5、可视化: 使用Scanpy的pl.pca函数对降维后的数据进行可视化。
import scanpy as sc
# 读取单细胞数据
adata = sc.read_10x_mtx('path/to/data', var_names='gene_symbols', cache=True)
# 预处理数据
sc.pp.filter_genes(adata, min_cells=3)
sc.pp.normalize_per_cell(adata, counts_per_cell_after=1e4)
sc.pp.log1p(adata)
# 进行降维
sc.tl.pca(adata)
# 聚类
sc.tl.louvain(adata)
# 可视化
sc.pl.pca(adata, color='louvain')
# 基因表达分析
# 基于聚类结果进行基因表达分析
sc.tl.rank_genes_groups(adata, 'louvain')
# 绘制基因表达差异热图
sc.pl.rank_genes_groups_heatmap(adata)
聚类后,数据可以进行下一步的分析
1、可视化聚类结果,使用工具如 t-SNE, UMAP, PCA 等降维方法可视化聚类结果,帮助我们更好的理解细胞间的关系。
2、基因表达分析,研究不同聚类组之间的基因表达差异,可以使用工具如 edgeR, limma, DEseq2 等。
3、转录因子关系网络分析,研究转录因子和基因之间的关系,可以使用工具如 scGRN, Monocle-CNV, Seurat-TFBS 等。
4、细胞周期和分化分析,研究细胞在周期和分化过程中的表达变化,可以使用工具如 CellcycleScoring, Monocle-CNV, Seurat-DIF 等。
5、疾病相关分析,研究聚类结果与疾病相关性,可以使用工具如 scDD, SCDE, Seurat-Disease 等
注:输入数据应该是一个10X格式的矩阵文件,这种文件格式通常包含三个部分: 一个稀疏矩阵文件,一个基因名称文件和一个细胞名称文件。
如果您没有单细胞数据,可以在网上找到一些公共数据集来进行测试,例如,可以在https://support.10xgenomics.com/single-cell-gene-expression/datasets/ 找到一些公共的10X数据集.
adata = sc.read_10x_mtx(path_to_data,var_names=‘gene_symbols’, cache=True)