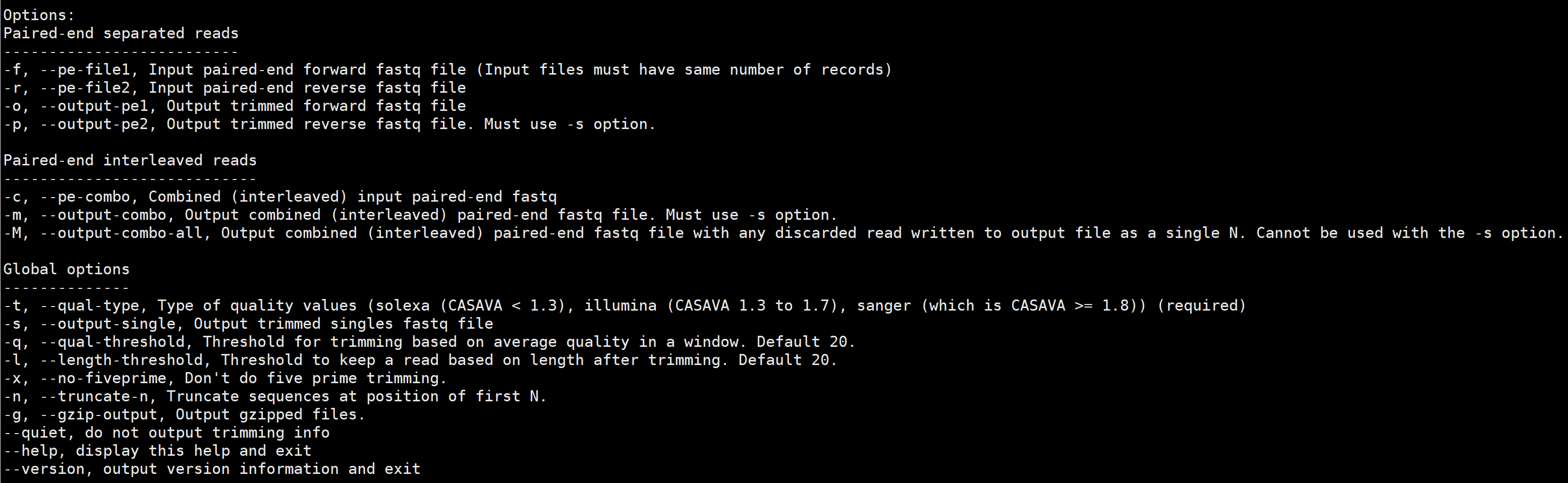
大多数现代测序技术产生的3 '端和5 '端质量降低的reads,这两个区域错误地calling base会对组装、下游生物信息学分析造成影响。sickle使用滑动窗口沿着质量和长度阈值,根据质量是否低于阈值来修剪reads的3 '端, 根据质量是否超过阈值来修剪reads的5 '端,还可以根据长度阈值丢弃reads。
sickle支持三种类型测序的质量值:Illumina、Solexa和 Sanger。
sickle修剪转录组测序fastq 5’和3’ reads 实例:
# 后台下载SRR3498212.sra
nohup prefetch SRR3498212 &
# 拆分sra为fastq
fastq-dump SRR3498212
# -f 输出fastq文件
# -t fastq测序碱基数据类型
# -o 输出修剪后fastq文件
# -q 质量阈值
# -l 长度阈值
sickle se -f SRR3498212.fastq -t sanger \
-o trimmed_SRR3498212.fastq -q 35 -l 45
# FastQ records kept: 34475799
# FastQ records discarded: 8018698
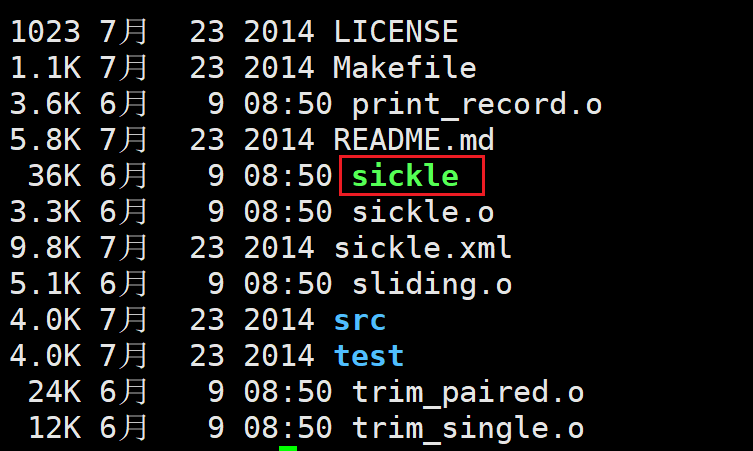
1. sickle安装
# 下载zip安装包
wget https://github.com/najoshi/sickle/archive/refs/tags/v1.33.zip
# 解压
unzip v1.33.zip
# 编译
cd sickle-1.33 && make
# 加入环境变量
echo 'export PATH=/path/sickle-1.33/:$PATH' >> ~/.basbrc
source ~/.bashrc
# 查看帮助
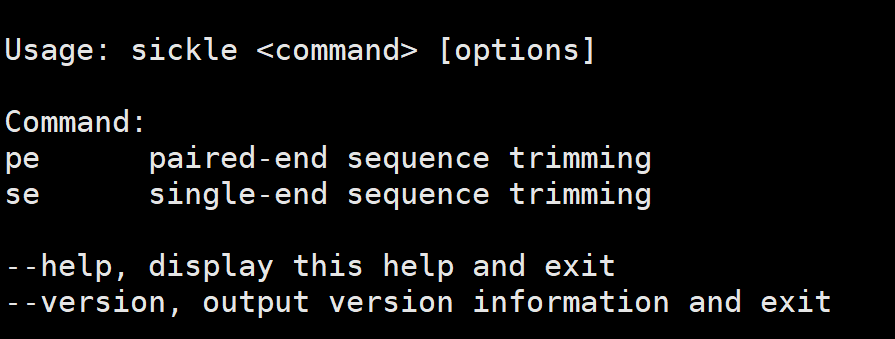
sickle -h
2. 单端测序数据修剪
sickle se获取一个输入单端fastq文件,并输出一个修剪后的fastq文件。 它还可以选择更改长度和质量用于微调的阈值,以及禁用5 '微调和启用N碱基截短序列。
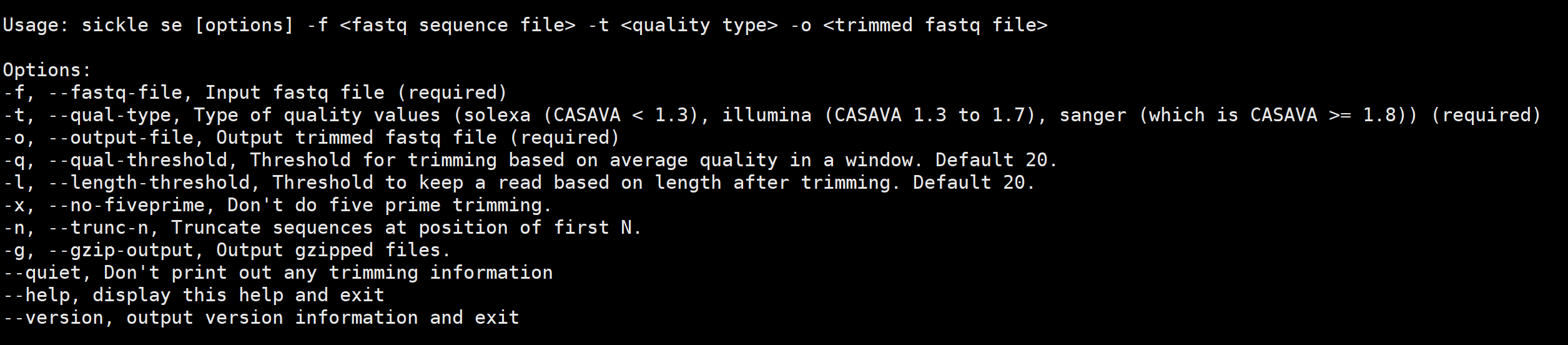
# -t 指定输入fastq质量类型为illumina
sickle se -f input_file.fastq -t illumina -o trimmed_output_file.fastq
# -q 指定质量阈值为33, -l 指定长度阈值为40
sickle se -f input_file.fastq -t illumina -o trimmed_output_file.fastq -q 33 -l 40
# -x 不进行5'端修剪reads, -x 第一个N碱基位置修剪序列
sickle se -f input_file.fastq -t illumina -o trimmed_output_file.fastq -x -n
# -g 输出.gz fastq文件
sickle se -t sanger -g -f input_file.fastq -o trimmed_output_file.fastq.gz
3. 双端测序数据修剪
sickle pe可以使用两种类型的输入进行操作。 首先,可以将两个双端文件作为输入,并输出两个修剪后的双端文件以及“singles”文件。 第二种形式以单个reads的组合输入文件。
“singles”文件包含正向或反向通过筛选器的reads方向。通过选项(-M), 可生成一个交错输出文件,其中任何未通过的reads过滤器将输出为一个FastQ记录与一个单一的“N”(其质量值是基于质量类型的最低可能值)。 可以更改长度以及用于修剪的质量阈值,以及禁用5 '-修剪, 允许用N碱基截短序列。
# -o 输出修剪的fastq1, -p 输出修剪会的fastq2, -s 输出singles文件
sickle pe -f input_file1.fastq -r input_file2.fastq -t illumina \
-o trimmed_output_file1.fastq -p trimmed_output_file2.fastq \
-s trimmed_singles_file.fastq
# 加入修剪质量和长度阈值
sickle pe -f input_file1.fastq -r input_file2.fastq -t illumina \
-o trimmed_output_file1.fastq -p trimmed_output_file2.fastq \
-s trimmed_singles_file.fastq -q 12 -l 15
# 加入N碱基修剪
sickle pe -f input_file1.fastq -r input_file2.fastq -t illumina \
-o trimmed_output_file1.fastq -p trimmed_output_file2.fastq \
-s trimmed_singles_file.fastq -n
# -c 单个组合的fastq文件作为输出
sickle pe -c combo.fastq -t sanger -m combo_trimmed.fastq \
-s trimmed_singles_file.fastq -n
sickle pe -t sanger -g -f input_file1.fastq -r input_file2.fastq \
-o trimmed_output_file1.fastq.gz -p trimmed_output_file2.fastq.gz \
-s trimmed_singles_file.fastq.gz
sickle pe -c combo.fastq -t sanger -M combo_trimmed_all.fastq
生信软件文章推荐
生信软件1 - 测序下机文件比对结果可视化工具 visNano
生信软件2 - 下游比对数据的统计工具 picard
生信软件3 - mapping比对bam文件质量评估工具 qualimap
生信软件4 - 拷贝数变异CNV分析软件 WisecondorX
生信软件5 - RIdeogram包绘制染色体密度图
生信软件6 - bcftools查找指定区域的变异位点信息
生信软件7 - 多线程并行运行Linux效率工具Parallel
生信软件8 - bedtools进行窗口划分、窗口GC含量、窗口测序深度和窗口SNP统计
生信软件9 - 多公共数据库数据下载软件Kingfisher
生信软件10 - DNA/RNA/蛋白多序列比对图R包ggmsa
生信软件11 - 基于ACMG的CNV注释工具ClassifyCNV
生信软件12 - 基于Symbol和ENTREZID查询基因注释的R包(easyConvert )
生信软件13 - 基于sambamba 窗口reads计数和平均覆盖度统计
生信软件14 - bcftools提取和注释VCF文件关键信息
生信软件15 - 生信NGS数据分析强大的工具集ngs-bits
生信软件16 - 常规探针设计软件mrbait
生信软件17 - 基于fasta文件的捕获探针设计工具catch
生信软件18 - 基于docker部署Web版 Visual Studio Code
生信软件19 - vcftools高级用法技巧合辑
生信软件20 - seqkit+awk+sed+grep高级用法技巧合辑
生信软件21 - 多线程拆分NCBI-SRA文件工具pfastq-dump









![YOLOv8_obb的训练、验证、预测及导出[旋转目标检测实践篇]](https://img-blog.csdnimg.cn/direct/c15101450fb647168e1d1e58b999ff45.png)