参考学习:传统蛋白质序列比对算法 - 知乎 (zhihu.com)
一、蛋白序列同源、相似
同源”(homology)和“相似”(similarity):
同源是指有相同的祖先,在这个意义上,无所谓同源的程度,两条序列要么同源,要么不同源。
而相似则是有程度的差别,如两条序列的相似程度达到30%或60%。一般来说,相似性很高的两条序列往往具有同源关系。【但也有例外,即两条序列的相似性很高,但它们可能并不是同源序列,这两条序列的相似性可能是由随机因素所产生的,这在进化上称为“趋同”(convergence),这样一对序列可称为同功序列。】
二、序列对比
序列比对算法主要分为全局比对(Global alignment)和局部比对(Local alignment)两种,分别从整体序列和局部序列来反映蛋白序列的特征。在实际上,生物序列只是局部相似而不是全长相似,我们往往采用局部比对算法,并且局部比对具有更高的灵敏性也更有生物意义。
序列比对分为序列两两比对和多序列比对。
首先在这里引入替换计分矩阵(打分矩阵)(替换矩阵)的概念,氨基酸序列的替换计分矩阵主要是考虑在进化过程中,不同氨基酸的替代对蛋白质功能和结构的影响不同,所以用相同氨基酸匹配得1分【等价矩阵】这种方法显然不行。
替换计分矩阵【氨基酸替换矩阵】:
氨基酸序列常用的打分矩阵主要有BLOSUM score matrix【blocks substitution matrix】,PAM score matrix,位置特异性矩阵PSSM。
【1】BLOSUM 矩阵
BLOSUM 矩阵是通过关系比较远的序列来获得矩阵元素的,BLOSUM 矩阵最早由 Steven Henikoff. 和 J.G Henikoff 在他们的论文中被提出。其中,他们从 BLOCKS 数据库中对那些在高度保守序列中的蛋白质家族进行观察测量进而整理出了氨基酸替换的概率。BLOSUM 打分矩阵的内容皆由观察得出。
BLOSUM 替换计分矩阵是一个log-odds矩阵, 基于序列之间的identity(大于一个阈值)将这些蛋白质序列cluster为500个group,每个group里面的序列做多序列比对,将保守无gap【空位】的区域划分为block,一共2000多个。identity可以取很多值,BLOSUM62矩阵用的是identity大于等于62%。然后基于这些block,可以找出20种氨基酸所有替换情况的频率。

计算过程如下:
一个氨基酸被另一个氨基酸替换所观察到的频率除以它俩因为随机而出现在一起的概率,然后取log值。
- 计算出一个block中每一列每一种配对出现的频数
- 遍历block的每一列,将特定配对情况的频数都加起来
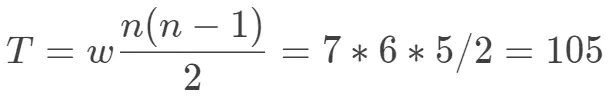
其中,w为列数,n为行数。
- 计算出每一种配对情况的观察到的频率
以AB配对为例:
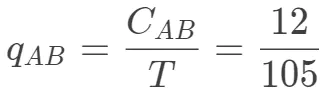
- 基于block计算某种氨基酸出现的概率
以A为例,AA配对贡献两个A,A(其他氨基酸)这类配对贡献一个,氨基酸在配对过程中总的出现次数是105*2
拓展到其它情况,氨基酸i出现的概率:
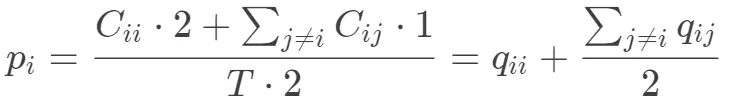
- 计算由于随机因素两个氨基酸一起出现的期望概率
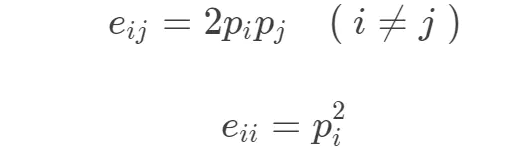
- 求出log odds ratio
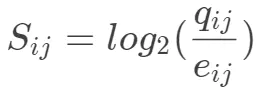
- 确定计分矩阵
上面的值再乘以2四舍五入取整即可。BLOSUM62矩阵广泛应用于双序列比对,也是BLAST程序默认调用的计分矩阵。BLOSUM62.txt
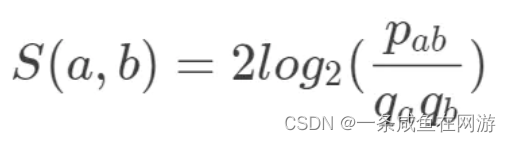
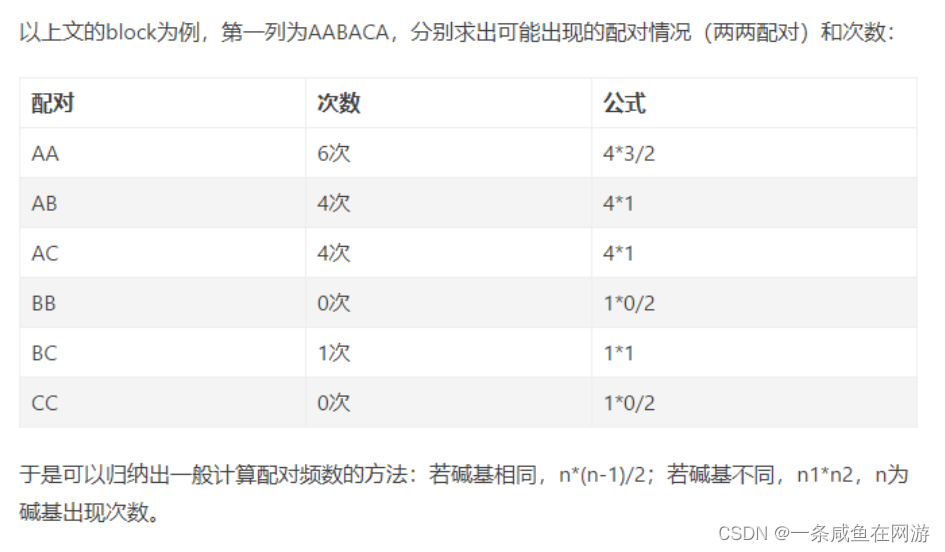
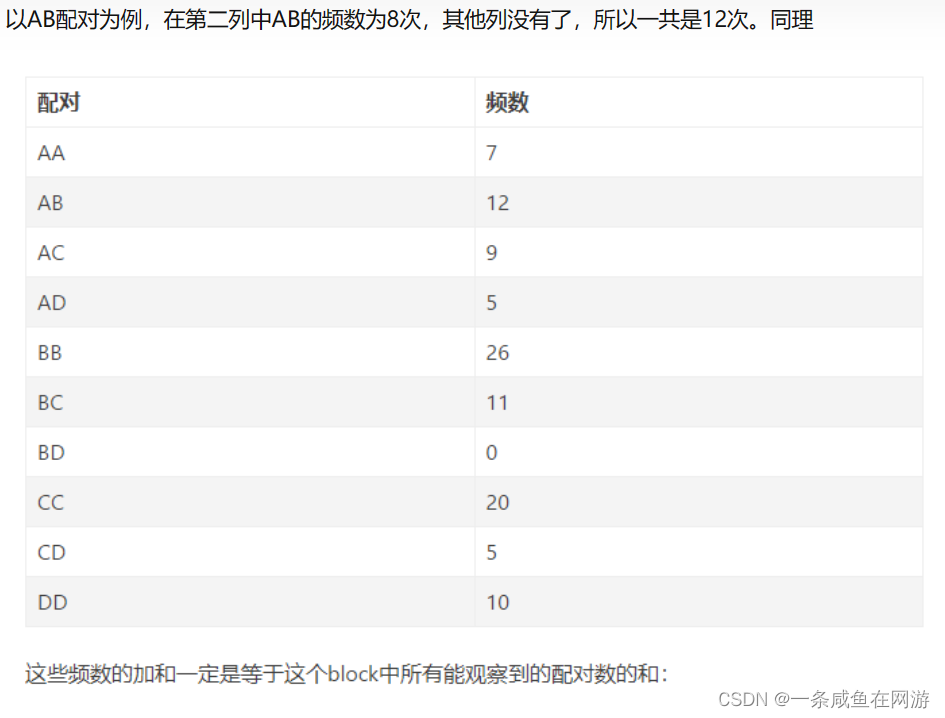
【2】PAM score matrix
【3】位置特异性矩阵PSSM
见参考学习链接:传统蛋白质序列比对算法 - 知乎 (zhihu.com)
对比野生型和突变型蛋白的blosum62【Sbl】:获得蛋白序列特征之一的方法




![《Python程序设计(第3版)》[美] 约翰·策勒(John Zelle) 第 9 章 答案](https://img-blog.csdnimg.cn/de24d16fb1c3466ba833ce4af4eb6d9a.png)