小罗碎碎念
先说明,目前小罗只是硕士,以下个人观点很有可能不准确,欢迎批评指正!!小罗虚心听取有益建议!!
众所周知,医学数据相比于其他领域的数据来说,属于小样本数据。那么从工科角度出发,模型的预测效果要想更精准,那么数据量就要尽可能大。好的,既然要解决的问题已经明确了,那么怎么解决呢?
如果是小罗的老粉,应该很快就能想到部分答案,例如多模态/多组学——模拟实际的临床场景,把病史、病理、影像还有测序的数据都纳入进来,这样数据量就上来了,但是问题又来了,不同模态的数据之间如何对准?模态缺失的问题如何解决?提取出来的高维特征如何降维、如何解释,都是需要考虑的问题。
既然上面说了,多模态多组学只是部分答案,那么肯定就还有其他的路子——跨癌种分析。先别急着否定,觉得选择这个方向就是扯淡。我们步子不要跨太大,不要一下就纳入几十种癌种,那样很难在短时间内得到有用的价值(因为肿瘤具有异质性)。我们可以先试着把具有关联的癌种分析一下,两三种就行(例如头颈癌、食道癌……)你试着想象一下,如果真的能找到一个泛化性尽可能大的方案,那你能走到哪个层次可就上不封顶了。另外,免疫治疗是一个很好的切入点,强烈建议大家提前布局,收集临床数据。
以上都属于偏临床的解决方案,那是属于在源头解决问题,其实工科方面也能缓解数据量小的问题——数据增强&交叉验证。这个我就不展开讲了,但是我会在后续的文章中把这些观点穿插进去,并且分享更多的项目复现经验。
(ps:现在知道小罗怎么确定每天的推文选题了吧,哈哈,目的导向非常明确滴)
2024-05-26|文献速递
交流群
欢迎大家来到【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。

一、泛癌种蛋白质基因组学:揭秘七大免疫亚型如何重塑癌症治疗

文献概述
这篇文章是一项关于肿瘤免疫的泛癌种蛋白质基因组学研究,它揭示了肿瘤免疫微环境中的多种免疫亚型,并探讨了它们与DNA变异、治疗反应和患者特征之间的关联。
主要发现:
- 研究者通过对超过
1000个肿瘤样本的蛋白质基因组学分析,发现了七种免疫亚型,这些亚型跨越了十种癌症类型。 - DNA变异与免疫亚型相关,并影响蛋白质组学特征。
- 免疫亚型中的激酶激活表明了潜在的治疗靶点。
- 数字病理学揭示了与免疫亚型相关的细胞浸润。
研究背景:
肿瘤微环境(TME)是癌症生态的关键组成部分,其中免疫和基质细胞可以以依赖于上下文的方式促进或限制恶性状态。- 免疫治疗在癌症治疗中的成功案例表明,患者的免疫系统可以被利用来对抗癌症。
研究方法:
- 使用了来自临床蛋白质组肿瘤分析联盟(CPTAC)的泛癌种蛋白质基因组数据。
- 该数据集包括基因组、转录组、表观基因组和蛋白质组数据。
- 采用了新的算法来剖析肿瘤微环境,并关联其功能和临床结果。
结果:
- 研究确定了基于细胞类型组成和通路活性的七个不同的免疫亚型。
- 深入分析了每个亚型独特的基因组、表观遗传、转录组和蛋白质组变化。
- 利用深度磷酸蛋白质组数据,研究了不同免疫亚型中的激酶活性,揭示了已知和潜在的新靶点。
- 通过机器学习算法应用于数字病理学图像,展示了免疫亚型与淋巴细胞浸润之间的相关性。
重点关注
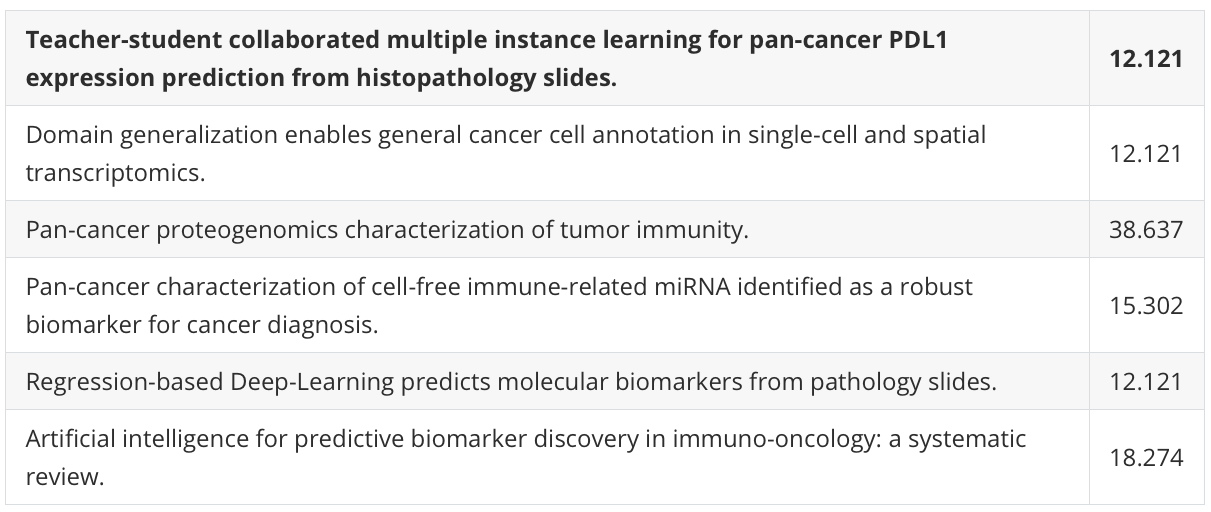
Figure 1 展示了免疫亚型的推导过程以及它们与肿瘤细胞、免疫细胞和基质细胞分数之间的关系。
以下是对图表的分析:
A部分 描述了免疫亚型的推导流程:
- 首先,基于蛋白质组学和RNA测序的多组学去卷积分析被执行,以估计每个肿瘤中的细胞类型组成。
- 同时,基于蛋白质组学的免疫相关通路得分被聚类,以定义10个免疫通路模块。
- 最后,将估计的细胞类型分数和10个免疫模块得分整合起来,将肿瘤聚类为不同的免疫亚型。
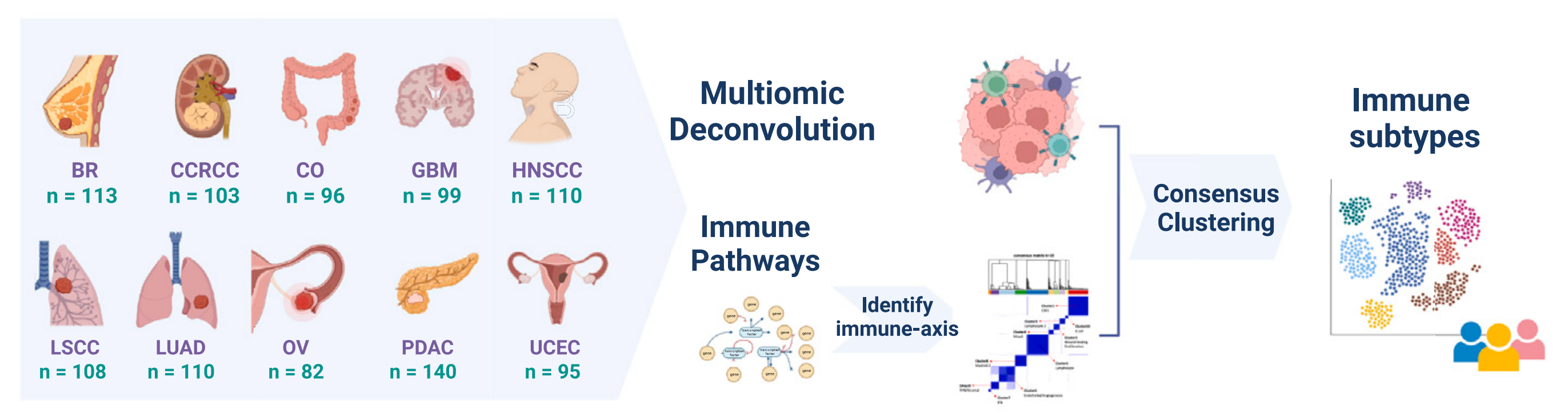
B部分 展示了一个热图,显示了每种癌症中肿瘤细胞百分比、免疫和基质得分以及细胞类型分数的平均值。显著差异水平(Bonferroni校正后的p值 < 1%)用星号(*)标出。
C部分 展示了每种癌症中细胞类型分数与生存结果之间的关联。热图展示了来自Cox比例风险回归模型的p值(符号化的对数10尺度)。在10%的FDR水平上显著的关联用黑点显示。
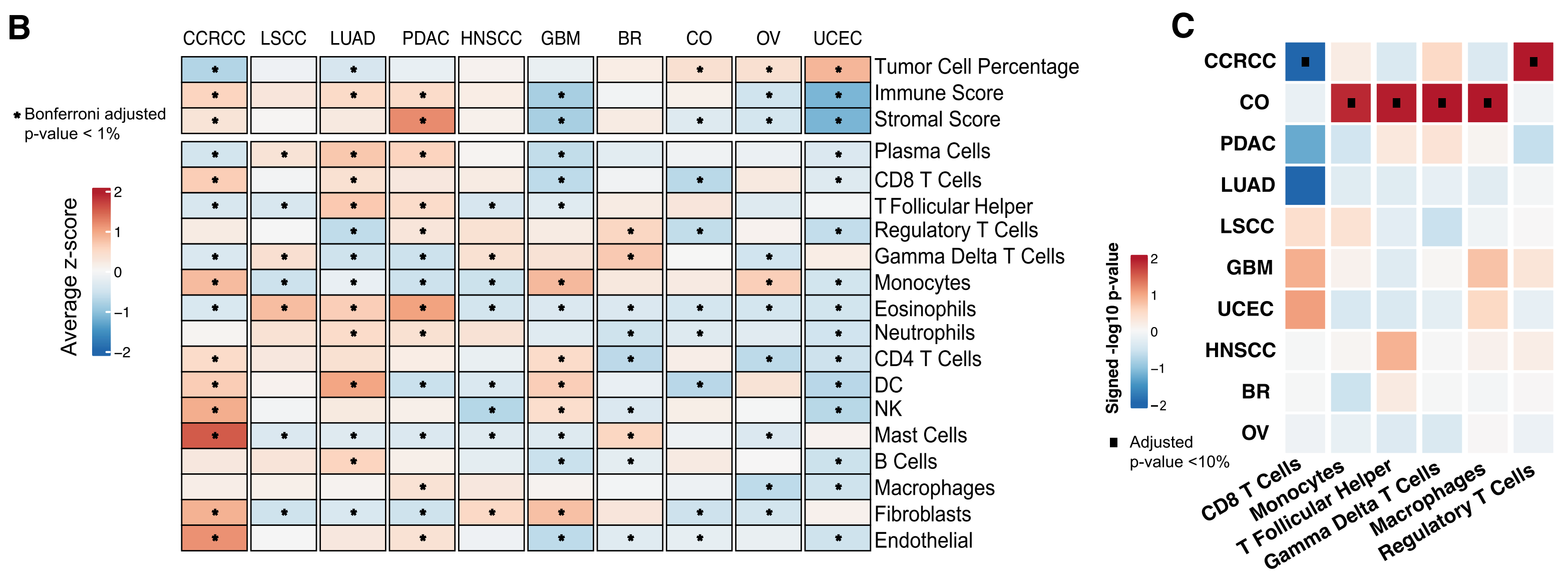
D部分 展示了Kaplan-Meier曲线,显示了每种癌症中CD8 T细胞分数与患者生存结果之间的特定癌症关联。对于每种癌症,使用第一和第三四分位数分别得出高和低CD8 T细胞分数的肿瘤。报告了logrank检验的p值。
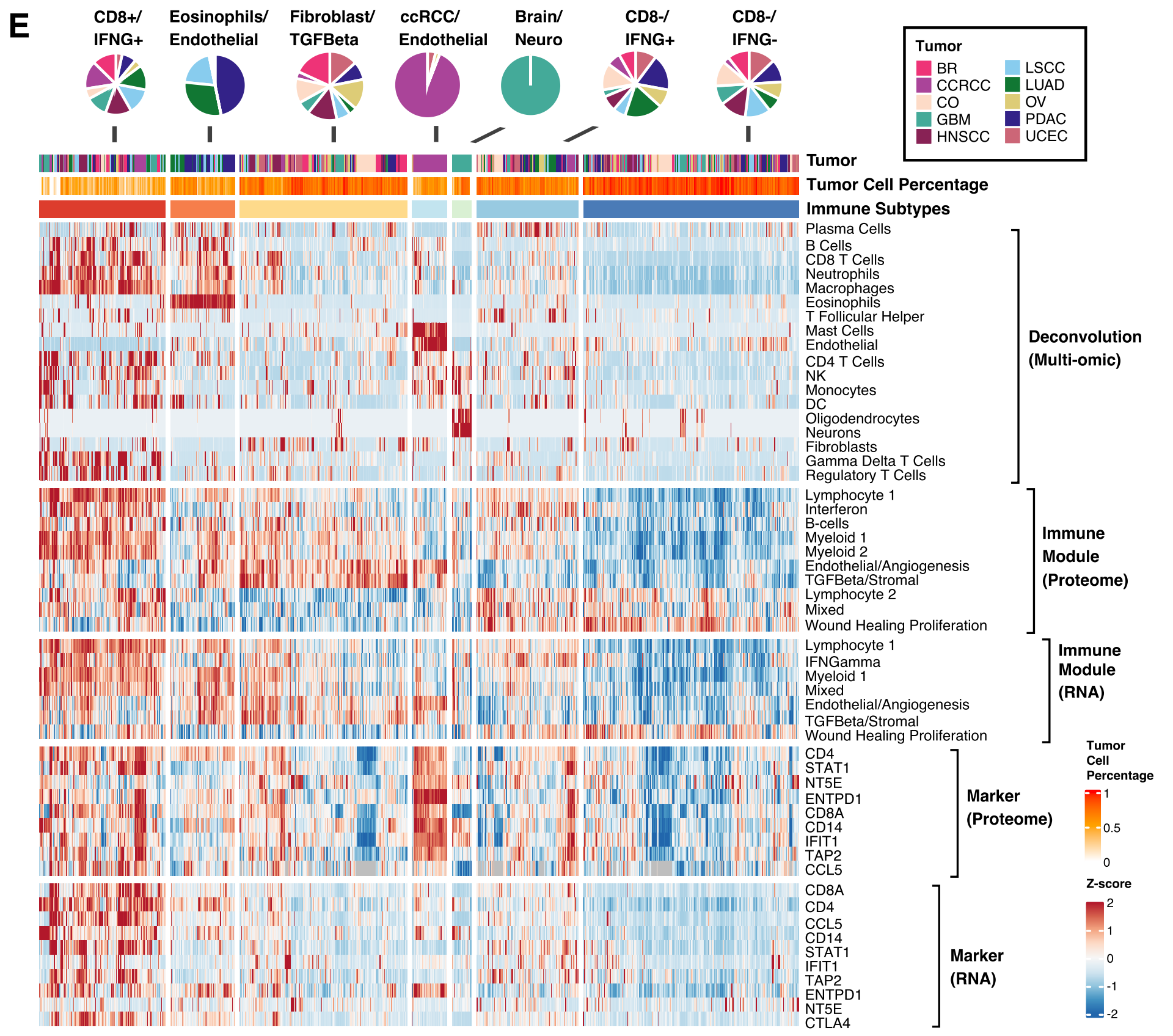
E部分 展示了一个热图,从上到下依次为:
- 通过去卷积分析估计的细胞类型分数。
- 和3. 分别基于蛋白质组和RNA的免疫模块通路得分。
- 和5. 基于细胞类型标记的蛋白质和RNA表达。
顶部的注释轨迹和饼图显示了不同肿瘤在免疫亚型中的分布。
F部分 展示了一个条形图,显示了每种癌症中分配到不同免疫亚型的样本比例。
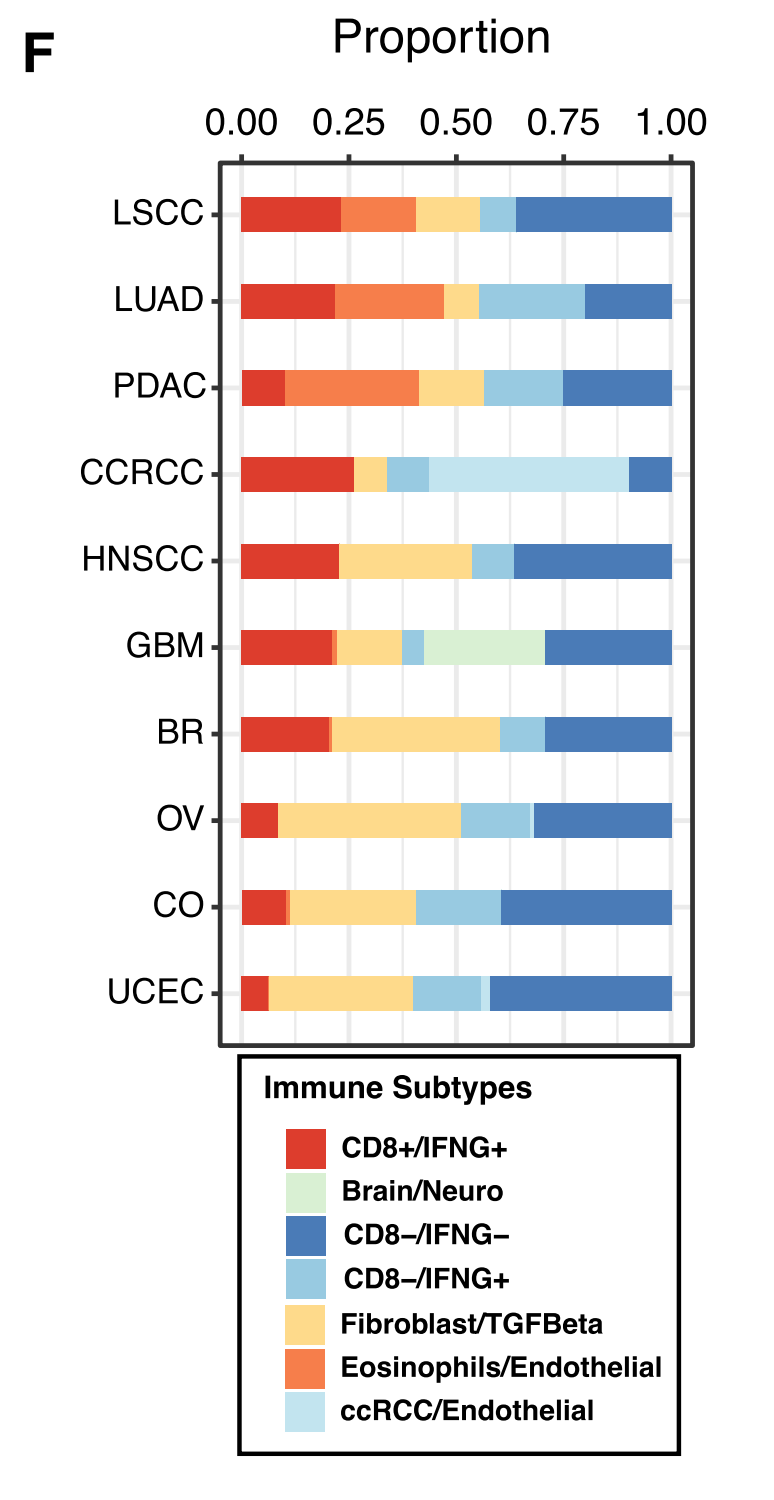
整体来看,Figure 1 揭示了免疫亚型是如何基于肿瘤的细胞组成和免疫通路活动被定义的,并且展示了这些亚型与不同癌症类型、患者生存结果以及CD8 T细胞分数的关联。通过这些分析,研究者能够更好地理解肿瘤微环境中的免疫细胞如何影响癌症的发展和治疗反应。
二、AI解码癌症免疫疗法:预测生物标志物

文献概述
这篇文章是关于在免疫肿瘤学领域使用人工智能(AI)进行预测生物标志物发现的系统综述。
文章首先指出免疫检查点抑制剂(ICIs)的广泛使用已经彻底改变了多种癌症的治疗方式,但选择可能从ICI中受益的患者仍然是一个挑战。AI方法允许在研究和精准免疫肿瘤学的发展中利用高维度的肿瘤学数据。
研究者们进行了一个系统的文献回顾,涵盖了研究ICI在癌症患者中的疗效预测的同行评审原创文章,这些研究涵盖了五种数据模式:
- 基因组学(包括基因组学、转录组学、表观基因组学)
- 放射组学
- 数字病理学(病理组学)
- 真实世界数据
- 多模态数据
在90项研究中,有80%的研究是在2021-2022年间发表的。其中,37项研究包括基因组数据,20项为放射组学,8项为病理组学,20项为真实世界数据,5项为多模态数据。72%的研究使用了标准的机器学习方法(ML),22%使用了深度学习方法(DL),6%同时使用了ML和DL。非小细胞肺癌(NSCLC)是最常研究的癌症类型,占36%,其次是黑色素瘤(16%),25%的研究包括泛癌研究。
文章指出,尽管包括的研究大多数显示了AI在预测免疫疗法受益方面的前景,但尚无研究提供足够高级别的证据以立即改变实践。需要事先计划的前瞻性试验设计,以覆盖这些软件生物标志物的所有生命周期步骤,从开发和验证到整合到临床实践中。
文章还详细介绍了AI在免疫肿瘤学中的应用,包括用于预测ICI疗效的不同数据类型(如基因组学、转录组学、表观基因组学、放射组学、病理组学、真实世界数据和多模态数据)的AI方法,以及如何通过AI技术识别新的生物标志物和元生物标志物。
此外,文章讨论了AI模型的可解释性、模型的异质性、研究的标准化、协议和指南,以及模型的泛化能力和稳健性等挑战。最后,文章得出结论,AI的使用在发现预测ICIs疗效的生物标志物方面日益增加,但要将AI方法直接应用于临床实践还面临诸多挑战。
重点关注
Figure 1 描述了开发用于预测免疫治疗(IO)疗效的模型的一般步骤,并展示了针对不同数据类型常用的方法论。
整个工作流程包含三个主要步骤:
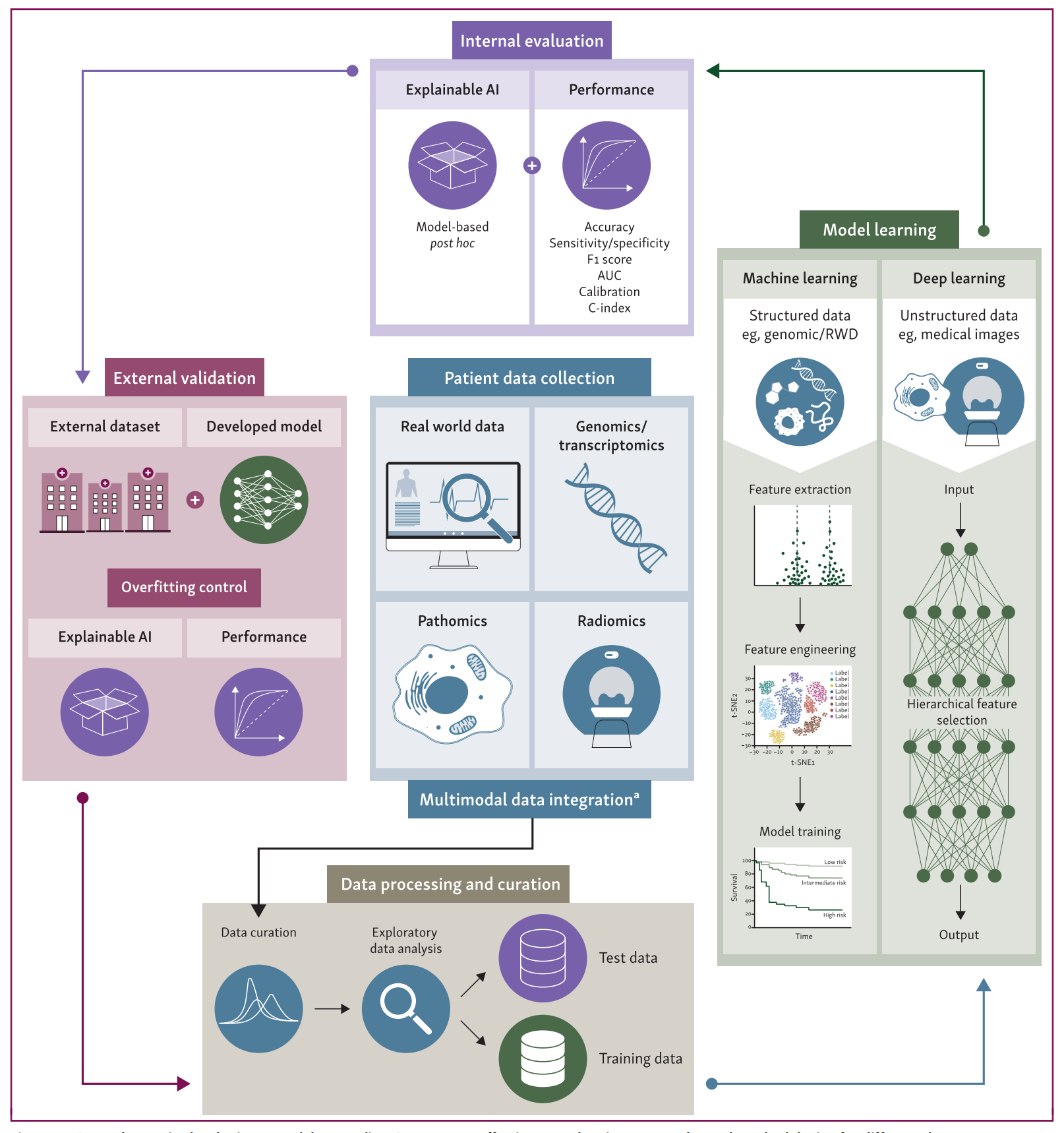
Step 1: 数据处理和整合
- 数据收集:涉及收集适当的组学数据(如基因组学、转录组学)、临床数据或图像数据。
- 数据存储:将收集的数据进行适当的存储和整合。
- 预处理:对数据进行预处理,以便于后续分析。
- 数据集划分:将处理后的数据分为训练集和测试集,用于模型的训练和评估。
Step 2: 模型学习
- 标准机器学习(ML):如果数据类型是结构化的(例如,真实世界数据和基因组数据),则适合使用标准ML。
- 深度学习(DL):主要用于图像数据(如数字病理学和放射组学)。
- 学习方式:根据最终目标和标记数据的可用性,选择有监督、半监督或无监督的学习方法。
Step 4 和 Step 5: 内部和外部验证
- 性能评估:在测试数据集上评估训练模型的性能,测试数据集包含“地面真实情况”。
- 模型解释:解释模型如何产生预测结果。
- 外部验证:在外部数据集上验证模型的预测能力和可解释性,以评估其在未见过的数据上的稳健性和普适性。
- 假设制定:根据内部和外部评估的结果,可以制定新的假设,以改进数据收集并训练改进的模型。
多模态数据整合
- 不同阶段整合:多模态数据整合可以在流程的不同阶段进行。
术语解释
- AI:人工智能
- AUC:曲线下面积(通常用于评估分类模型的性能)
- ML:机器学习
- RWD:真实世界数据
这个流程强调了在开发预测模型时,从数据的收集、处理、模型训练到验证评估的每个步骤的重要性。通过这种系统化的方法,可以提高模型的准确性和可靠性,进而在实际应用中更好地预测免疫治疗的疗效。
三、癌症检测新方向:一滴血中的微小RNA密码

文献概述
这篇文章是关于一种新型的、非侵入性的癌症检测方法的研究,主要聚焦于循环细胞外免疫相关微小RNA(cf-IRmiRNAs)的表达特征。
目的与方法:
- 研究旨在建立一种基于cf-IRmiRNAs的早期癌症检测标志物。
- 分析了来自
15,832名参与者的循环miRNA图谱,包括13种癌症类型和对照组。 - 数据随机分为训练集、验证集和测试集(比例为7:2:1),另有一个外部测试集包含684名参与者。
- 通过
最小绝对收缩和选择算子(LASSO)方法,从100个差异表达的cf-IRmiRNAs中筛选出39个。 - 使用
五种机器学习算法构建cf-IRmiRNAs标志物,其中基于XGBoost算法的分类器在验证集上显示出优异的癌症检测性能。
主要发现:
- XGBoost算法构建的分类器在验证集上(AUC: 0.984)和测试集上(AUC: 0.983)均表现出色。
- 该分类器还能成功检测早期癌症,特别是肺癌、前列腺癌和胃癌。
- 此外,该分类器还能区分良性和恶性肿瘤。
研究意义:
- 这项研究是迄今为止规模最大、最全面的关于cf-IRmiRNAs的泛癌分析。
- 提供了一种有前景的非侵入性诊断生物标记物,用于早期癌症检测,可能对临床实践产生影响。
结论:
- 尽管研究设计为回顾性,但cf-IRmiRNAs标志物显示出了在早期癌症检测中的高准确性和稳定性。
- 需要在未来进行大规模前瞻性和多中心试验以进一步验证。
重点关注
四、从分类到回归:如何让机器更懂病理,提升癌症患者生存预测准确性?

文献概述
这篇文章的标题是《Regression-based Deep-Learning predicts molecular biomarkers from pathology slides》,由 Omar S. M. El Nahhas 和 Jakob Nikolas Kather 等人撰写,发表在《Nature Communications》上。
文章的核心观点是提出了一种基于回归的深度学习方法,用于从病理幻灯片中预测分子生物标志物。研究团队开发并评估了一种自监督的、基于注意力机制的弱监督回归方法,该方法能够直接从 11,671 幅患者的病理图像中预测连续的生物标志物,涵盖了九种癌症类型。他们特别测试了多种临床和生物学上相关的生物标志物,包括同源重组缺陷(HRD)评分,这是一种临床上使用的泛癌生物标志物,以及肿瘤微环境中关键生物过程的标志物。
研究发现,使用回归方法显著提高了生物标志物预测的准确性,并且与分类方法相比,回归预测与已知临床相关区域的对应性更好。在结直肠癌患者的大型队列中,基于回归的预测评分比基于分类的评分具有更高的预后价值。
文章还讨论了数字病理学的发展,以及深度学习(DL)在预测病理诊断流程中的生物标志物方面的应用。研究指出,尽管大多数已发表的方法限于分类问题,但许多生物标志物的真实值是连续的,这导致在训练深度学习模型之前需要将这些值二值化或分类,从而造成信息丢失。因此,作者提出回归分析是解决这一问题更合适的方法。
研究结果表明,CAMIL(contrastively-clustered attention-based multiple instance learning)回归方法在预测 HRD 状态方面优于现有的分类方法和 Graziani 等人提出的回归方法。CAMIL 回归模型在多个癌症类型中实现了高 AUROC(Area Under the Receiver Operating Characteristic curve)值,并且在外部验证队列中表现出更好的泛化能力。
此外,文章还探讨了 CAMIL 回归方法在预测肿瘤细胞增殖、基质分数和免疫细胞相关生物标志物方面的性能,并与 CAMIL 分类方法和 Graziani 等人的回归方法进行了比较。在所有测试的癌症类型中,CAMIL 回归方法在 28 个模型中的 AUROC 超过 0.70,显示出其强大的预测能力。
最后,文章讨论了 CAMIL 回归方法在提高生物标志物预测的准确性和对应性方面的优势,并指出这种方法在计算病理学文献中填补了重要的空白。作者认为,他们的开源回归方法为连续生物标志物分析提供了一个有希望的替代方案,并可能对精准医疗领域产生重要影响。
重点关注
Fig. 1 展示了这项研究中的端到端实验工作流程的概览,包括图像预处理、建模、性能指标评估以及所使用的队列。
以下是对图的详细分析:
A. 图像预处理流程和瓦片级特征提取:
- 使用带有预训练的ImageNet权重的ResNet50模型,进行推理以提取特征。
- 结合检索对比聚类(RetCCL)模型,为每个患者生成一个特征矩阵。
B. 建模架构的描述:
- 利用基于注意力的多重实例学习(attMIL)应用于自监督提取的特征。
- 结构包含三个独立训练的头:
- 一个用于CAMIL分类。
- 一个用于按照Graziani等人提出的方法进行回归。
- 第三个用于本文介绍的CAMIL回归方法。
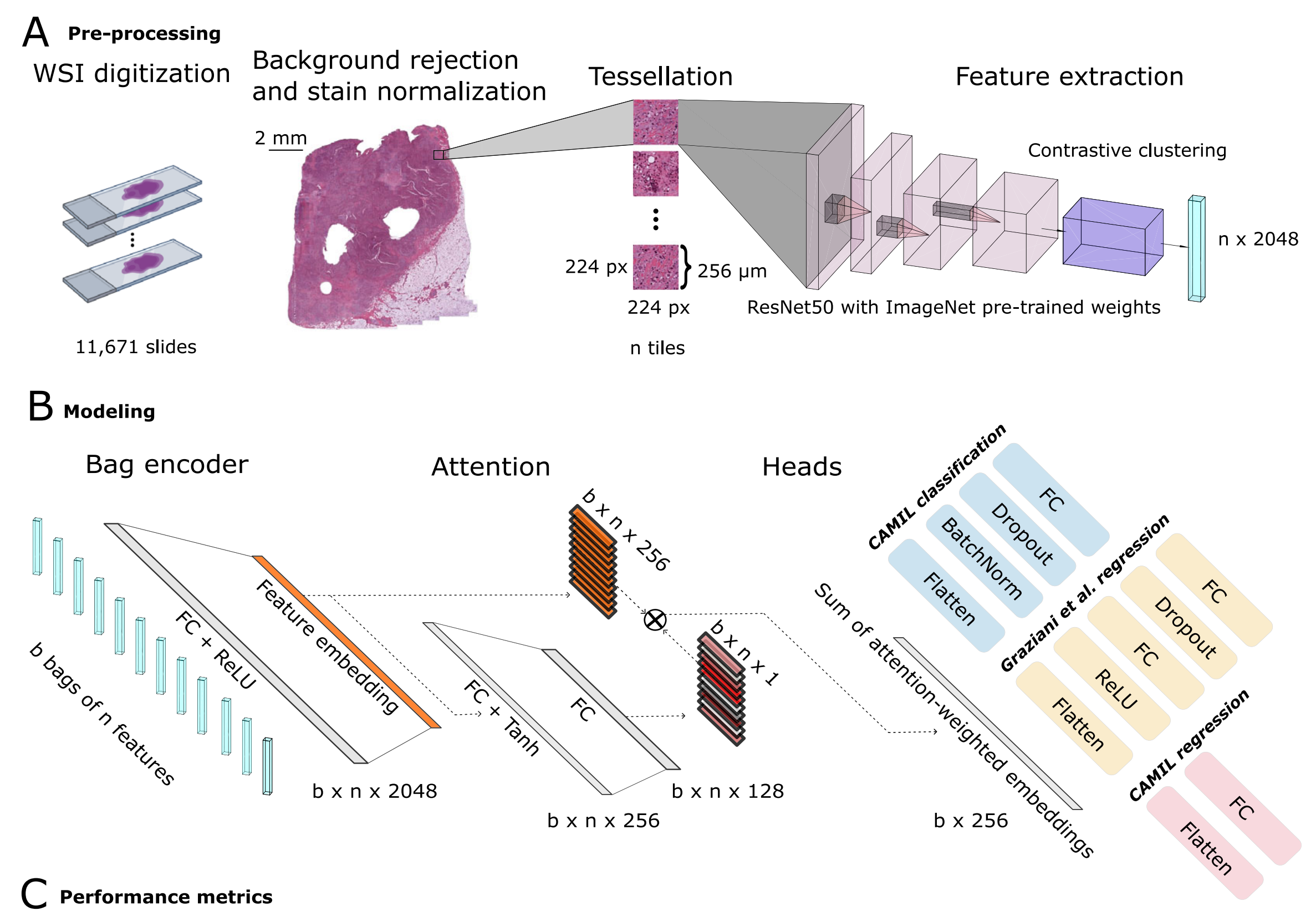
C. 性能指标及其相应的置信区间(CI):
- 评估模型三个独立训练头的性能指标包括:
- 对于回归模型,使用皮尔逊相关系数(Pearson’s r)。
- 对于所有模型,使用接收者操作特征曲线下面积(AUROC)。
- 对同源重组缺陷(HRD)和生物过程生物标志物,进行了配对双尾DeLong测试。
- 进行了注意力热图的专家评审,并结合单变量(UV)和多变量(MV)Cox比例风险(PH)模型,用于生物过程模型。
D. 所使用的队列的图表表示:
- 内圈和外圈分别表示用于训练和外部验证的队列。
- 训练队列来源于癌症基因组图谱(TCGA)项目中的所有临床目标。
- 外部验证队列来源于临床蛋白质组肿瘤分析联盟(CPTAC)的努力,以及针对HRD目标和生物过程生物标志物的Darmkrebs:Chancen der Verhütung durch Screening(DACHS)研究。
- 考虑的生物过程生物标志物包括肿瘤浸润淋巴细胞区域分数(TIL RF)、增殖(Prolif.)、白细胞分数(LF)、淋巴细胞浸润签名得分(LISS)和基质分数(SF)。
- 研究中考虑的癌症类型包括乳腺癌(BRCA)、结直肠癌(CRC)、胶质母细胞瘤(GBM)、肺腺癌(LUAD)、肺鳞状细胞癌(LUSC)、胰腺腺癌(PAAD)、子宫内膜癌(UCEC)、肝细胞癌(LIHC)和胃癌(STAD)。
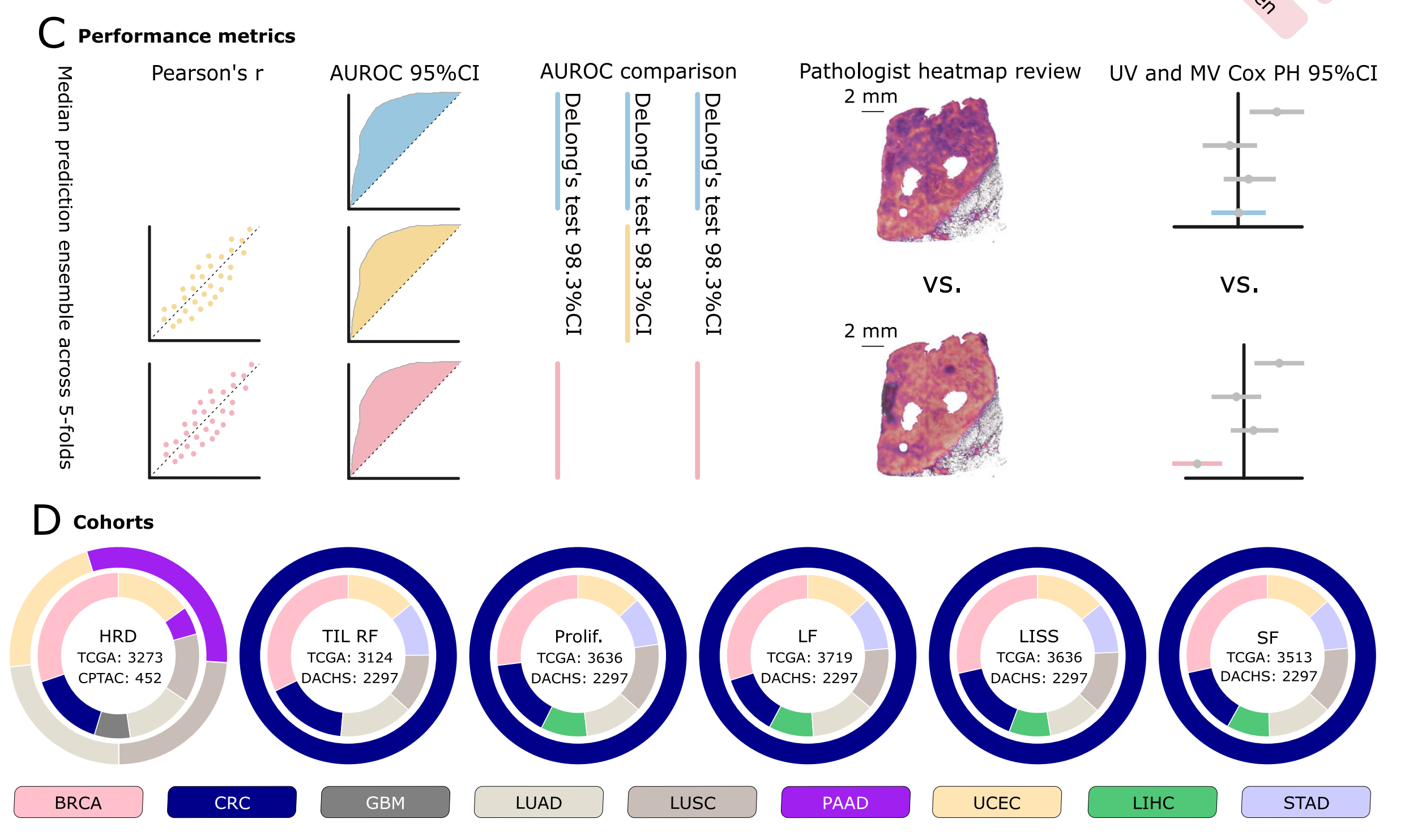
这个流程图详细说明了如何从原始的病理幻灯片图像中提取特征,并使用这些特征通过深度学习模型来预测生物标志物,以及如何评估这些模型的性能。此外,还展示了所使用数据的来源和队列的分配,这对于确保研究的透明度和可重复性至关重要。
五、从微观到宏观:Cancer-Finder算法精准识别恶性细胞

文献概述
这篇文章介绍了一种名为Cancer-Finder的新型深度学习算法,它基于领域泛化(domain generalization)来识别单细胞转录组测序(scRNA-seq)数据中的恶性细胞。Cancer-Finder通过在多个具有不同分布的数据集上训练,学习出一个泛化模型,能够直接区分未知数据集中的恶性和正常细胞。该算法在单细胞数据中的识别准确度平均达到了95.16%,并且能够扩展到空间转录组(spatial transcriptomics, ST)数据中,准确识别空间幻灯片上的恶性区域。
Cancer-Finder的开发背景是基于肿瘤异质性对有效癌症治疗的阻碍,scRNA-seq技术的发展让研究者能够在单细胞水平上全面理解肿瘤的异质性,从而推动个性化治疗的发展。ST技术作为scRNA-seq的补充,捕捉组织内部的转录活性的空间背景,越来越多地应用于癌症研究。在这些研究中,精确标注单个细胞或斑点(ST中的测量单位)的恶性状态是至关重要的。
Cancer-Finder通过一个深度神经网络实现,该网络由特征提取模块和分类模块组成。它利用风险外推(risk extrapolation)方法来进行领域泛化,优化模型以在所有组织中实现高准确度,并通过减少训练领域之间的风险差异来降低模型对广泛分布变化的敏感性。Cancer-Finder还集成了一个解释性模块,使用修改后的显著性图来研究区分恶性细胞和非恶性细胞的关键特征。
在对多个组织的scRNA-seq数据进行训练后,Cancer-Finder能够成功提取和转换新样本的scRNA-seq和ST数据的特征,推断细胞的恶性状态,并根据恶性状态直接对癌症数据中的细胞进行可视化。此外,Cancer-Finder在空间转录组数据上的应用表明,它能够扩展到包括不同癌症类型的数据,并在训练后达到高预测准确度。
最后,Cancer-Finder在分析透明细胞肾癌(ccRCC)的空间转录组数据时,成功识别了一个由10个基因组成的基因签名,这些基因在肿瘤-正常组织界面显著共定位和富集,并且与透明细胞肾癌患者的预后有很强的相关性。这表明Cancer-Finder是一个高效且可扩展的恶性细胞注释工具,将有助于使用单细胞和空间转录组数据发现生物学机制。
重点关注
Fig. 1 提供了 Cancer-Finder 及其应用的概述。
Cancer-Finder 是一个可扩展的框架,它利用单细胞测序数据来准确注释细胞的恶性状态,并且可以轻松扩展以注释其他数据类型(例如空间转录组数据,ST)。
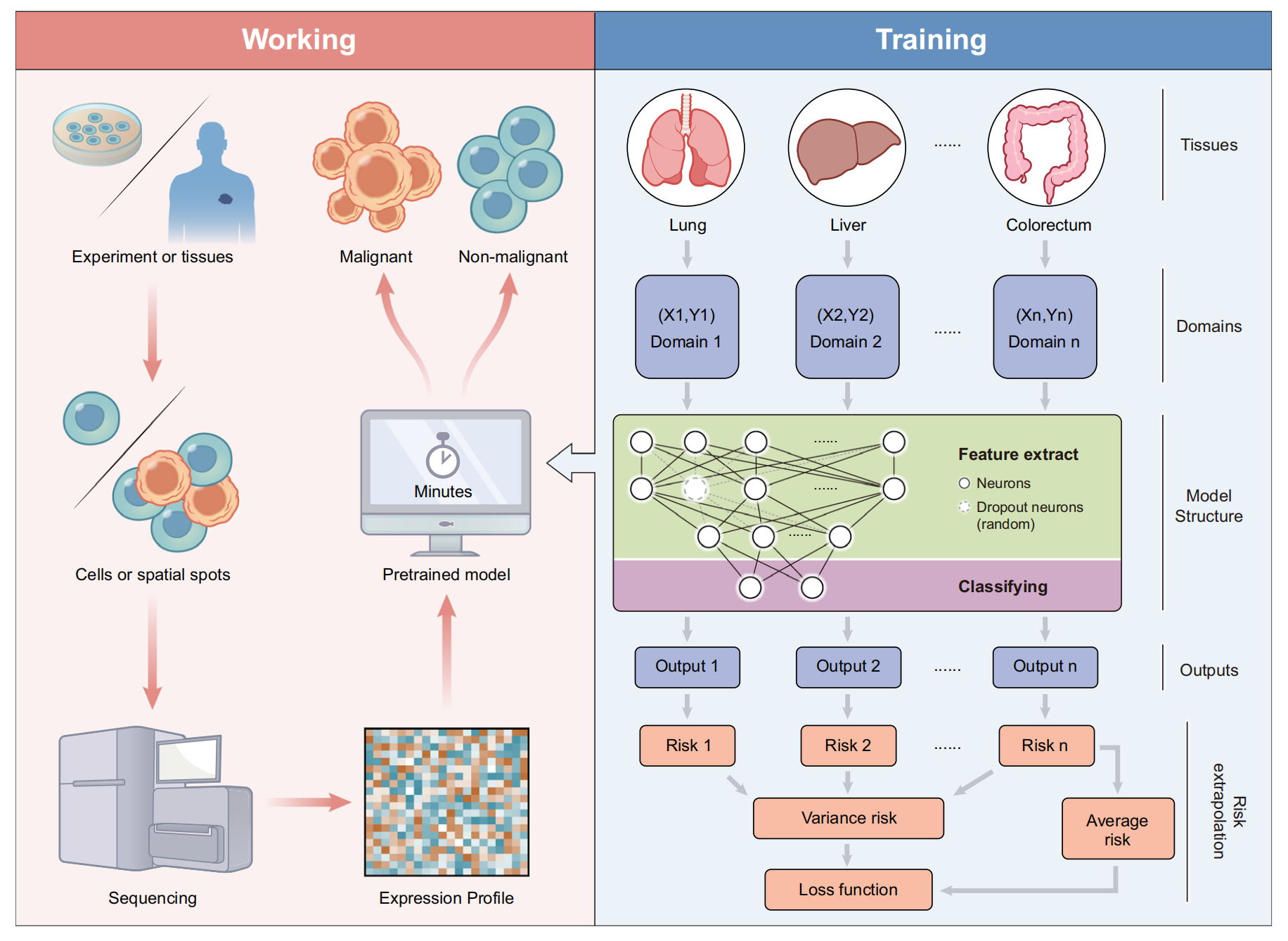
预训练模型能够准确且快速地识别来自癌组织的恶性细胞。为了抵消不同组织之间的差异,Cancer-Finder 采用了领域泛化训练策略来提高一般区分性能,并在未探索的领域中准确识别恶性细胞。通常,“领域”指的是模型训练时使用的具体数据类型或类别。在这里,作者将这一概念应用于单细胞或空间数据中细胞恶性状态的注释,假设来自不同组织的数据显示出不同的领域特征。
Cancer-Finder 的模型是一个由输入层、两个隐藏层(用于特征提取)和分类层组成的神经网络。它使用风险外推方法来进行领域泛化,这优化了模型在所有组织中的高准确度,因为减少训练领域之间的风险差异可以降低模型对广泛分布变化的敏感性。为了评估模型在多个领域中的表现,该方法最小化了两种类型的全局风险:方差风险和平均风险。
具体来说,Cancer-Finder 的工作流程可以概括为以下几个步骤:
- 预训练模型:使用单细胞测序数据预训练模型,使其能够识别恶性细胞。
- 领域泛化:通过训练策略,使模型能够处理来自不同组织(即不同领域)的数据,并保持高准确度。
- 特征提取:利用神经网络的隐藏层提取数据中的关键特征。
- 分类:基于提取的特征,模型对细胞是否为恶性进行分类。
- 风险最小化:通过最小化方差风险和平均风险,提高模型在不同领域中的泛化能力。
这种设计使得 Cancer-Finder 不仅能够处理单一类型的数据,还能够适应多种不同的数据源,包括空间转录组数据,从而在肿瘤微环境的研究中具有广泛的应用潜力。通过这种方式,Cancer-Finder 有助于推动个性化医疗和精准肿瘤治疗的发展。
六、一种用于全癌种PDL1表达预测的深度学习方法

文献概述
这篇文章的标题是“Teacher-student collaborated multiple instance learning for pan-cancer PDL1 expression prediction from histopathology slides”,作者Darui Jin等人,发表在《Nature Communications》上。文章介绍了一种用于全癌种PDL1表达预测的深度学习方法,该方法基于苏木精-伊红(H&E)染色的组织病理学幻灯片。
背景:
PDL1(Programmed cell death ligand 1)是一个重要的生物标志物,通常通过免疫组化(IHC)进行量化,但缺乏已建立的组织病理学模式。深度学习有助于组织病理学评估,但异质性和缺乏空间解析注释对精确分析构成挑战。
方法:
文章提出了一种弱监督学习方法,利用大量RNA测序(bulk RNA sequencing)来预测H&E幻灯片上的PDL1表达。该方法扩展了多实例学习(Multiple Instance Learning, MIL)范式,结合了教师-学生框架,通过动态伪标签处理幻灯片内异质性,并使用时间集成模型蒸馏检索未标记实例。
评估:
该方法在12,299张幻灯片上进行了评估,涵盖了20种实体瘤类型。在将PDL1作为已建立生物标志物的9种肿瘤中,该方法在新鲜冷冻和甲醛固定标本上的加权平均曲线下面积(AUC)分别为0.83和0.74。
结果:
该方法能够预测PDL1表达模式,并在20张幻灯片上通过IHC验证,提供了与PDL1相关的组织学见解。这表明深度学习在从H&E图像中识别分子变化的多样化组织学模式方面具有潜力。
讨论:
PDL1表达的预测基于H&E幻灯片是可能的,并且MILTS(教师-学生协作的多实例学习框架)能够捕获与分子变化相关的有意义的组织学模式。这些发现强调了人工智能在通过大量数字化组织病理学幻灯片及其注释来增强病理学家能力方面的效用。
结论:
文章展示了MILTS在预测一系列癌症中PDL1表达方面的有效性,并揭示了与PDL1表达相关的多样化组织学模式。这可能有助于减少额外测试的需求,并为病理学家提供更精确的解释免疫逃逸模式的见解。
文章还讨论了该方法的局限性,包括新鲜冷冻和甲醛固定组织切片之间的性能差异,以及PDL1表达水平与组织病理学预测能力之间的相关性。作者指出,未来的研究应该关注缩小这些差距,并提高使用单模态数据训练的模型的泛化能力。
重点关注
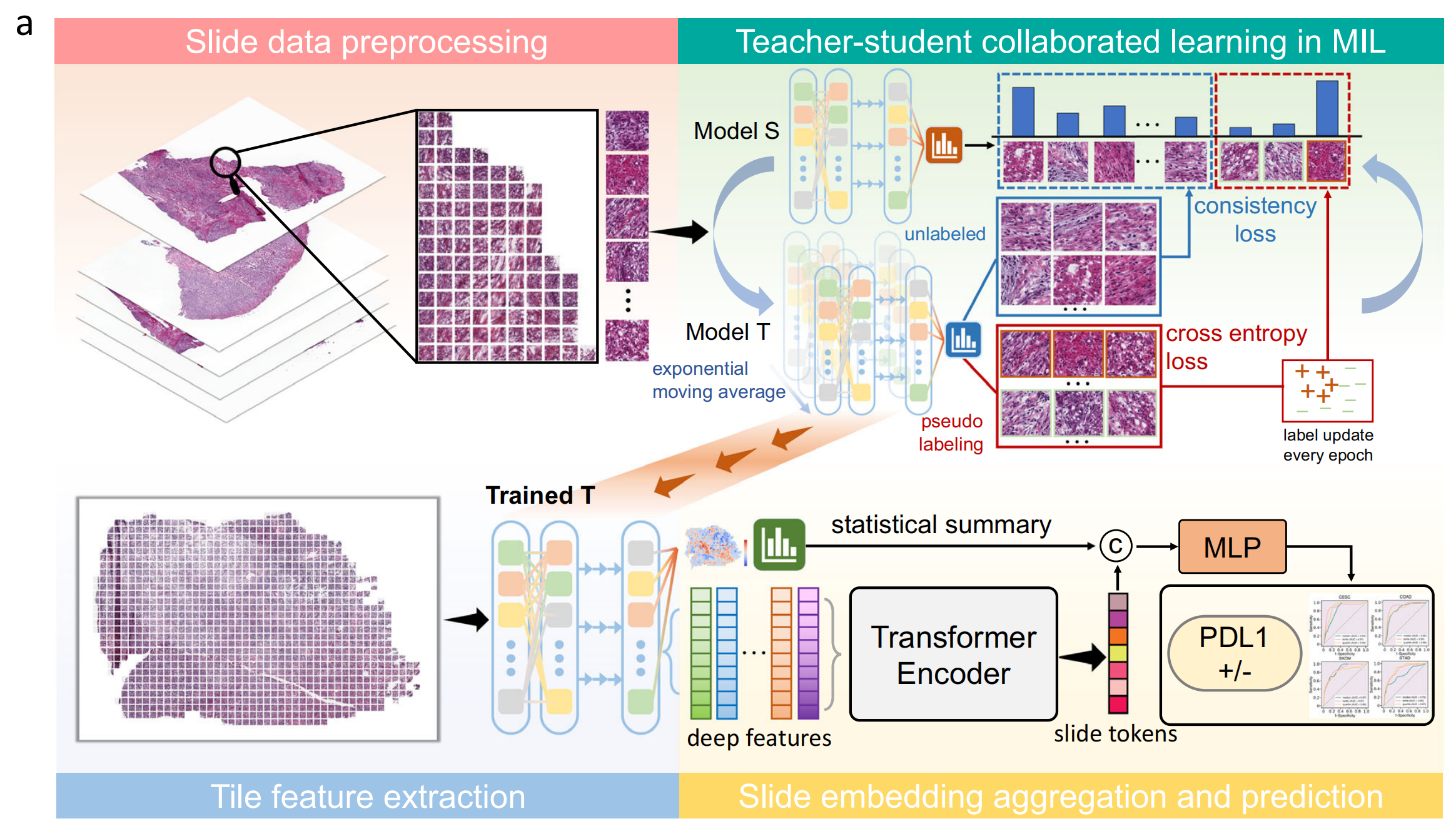
Fig. 1 展示了MILTS(教师-学生协作的多实例学习框架)的工作流程及其在临床上与PDL1相关的肿瘤上的性能表现。
以下是对图的详细分析:
a 部分描述了MILTS的训练和推理工作流程,分为三个主要步骤:
-
数据划分:首先,将患者队列的数据分为训练集、验证集和测试集。
-
图像处理:接着,对数据进行分块(patching)和随机增强(random augmentations),以增加数据集的多样性并提高模型的泛化能力。
-
网络训练与特征提取:然后,利用得到的图像块训练一个基于MIL的教师-学生协作网络。训练完成后,教师模型或学生模型作为特征提取器,提取统计特征和深度特征。同一幻灯片中的图像块的深度特征进一步融合成幻灯片标记(slide token),并与块级特征的统计摘要结合,用于训练一个多层感知器(MLP)分类器,该分类器提供患者级别的诊断。
- MIL:多实例学习,一种机器学习范式,其中每个实例(在这里是一个图像块)属于一个包含多个实例的“包”,并且整个包的标签用于训练。
- S:学生模型,学习从教师模型中获取的知识。
- T:教师模型,提供指导和伪标签。
- C:串联,将不同的特征组合在一起。
- MLP:多层感知器,一种前馈人工神经网络,用于分类任务。
b 部分展示了不同肿瘤类型的幻灯片数量,这可能与每种肿瘤类型的样本量或研究重点有关。
c 部分是一个图表,展示了MILTS模型在FFPE(甲醛固定石蜡包埋)幻灯片和新鲜冷冻幻灯片上的性能。性能通过曲线下面积(AUC)来衡量,这是一种评估分类模型性能的指标,特别适用于医学诊断领域。AUC值越高,模型的诊断能力越好。图表中可能还展示了不同肿瘤类型的性能,允许比较模型在不同类型肿瘤中的诊断效果。
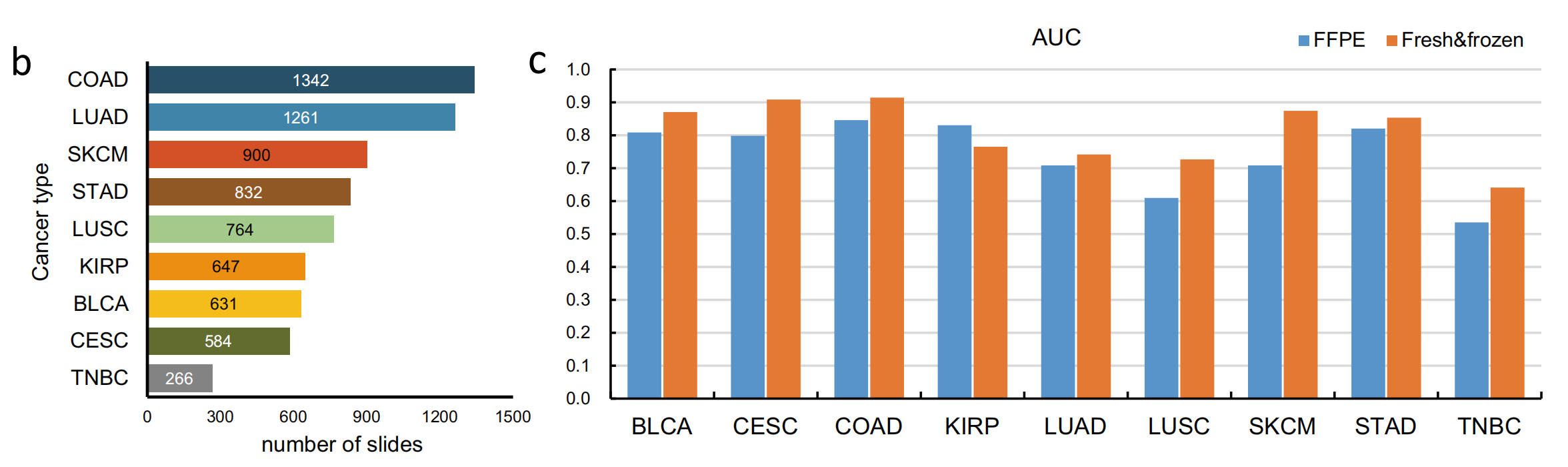
总的来说,Fig. 1 清晰地展示了MILTS模型的工作流程和性能评估,突出了其在不同肿瘤类型上的诊断潜力,并通过提供源数据支持了研究的透明度和可重复性。