文章《Density functional model of threshold voltage shifts at High-K/Metal gates》,是由R. Cao、Z. Zhang、Y. Guo、J. Robertson等人撰写,发表在《Solid-State Electronics》期刊上。通过密度泛函理论(Density Functional Theory, DFT)分析了在高K/金属栅极CMOS结构中用于设定阈值电压的氧化物偶极层。
1. 引言 (Introduction)
文章首先介绍了高K介质材料(如HfO2)在CMOS晶体管栅极结构中的应用,以及金属栅极的必要性,以最小化栅极电极耗尽深度。文章提出了两个关键问题:如何获得调整n型和p型MOSFET阈值电压(Vth)所需的两种不同的有效功函数,以及如何实现这些功函数的调整。
- 使用两种不同的金属:初始的方法是使用两种不同的金属来实现两种不同的工作函数,但这种方法由于费米能级钉扎(Fermi Level Pinning, FLP)或金属的过度反应性而变得不便。
- 使用金属氧化物偶极层:另一种方法是使用一种反应性较低的金属(如TiN)与金属氧化物偶极层(如La2O3或Al2O3)结合使用,通过调整每个电极的有效工作函数来实现所需的阈值电压调整。
文章还回顾了之前基于金属氧化物离子密度或电负性尺度的经验模型,并指出了这些模型的局限性。
2. 方法 (Methods)
文章详细描述了使用密度泛函理论(DFT)计算氧化物偶极层的偶极工作函数偏移的方法。具体包括:
- 使用VASP软件和平面波赝势进行计算。
- 使用PBE-GGA泛函进行能量最小化。
- 通过Heyd-Scuseria-Ernzerhof (HSE)混合泛函修正带隙。
- 计算了SrO、La2O3、HfO2和Al2O3的电子结构和带对齐。
3. 结果 (Results)
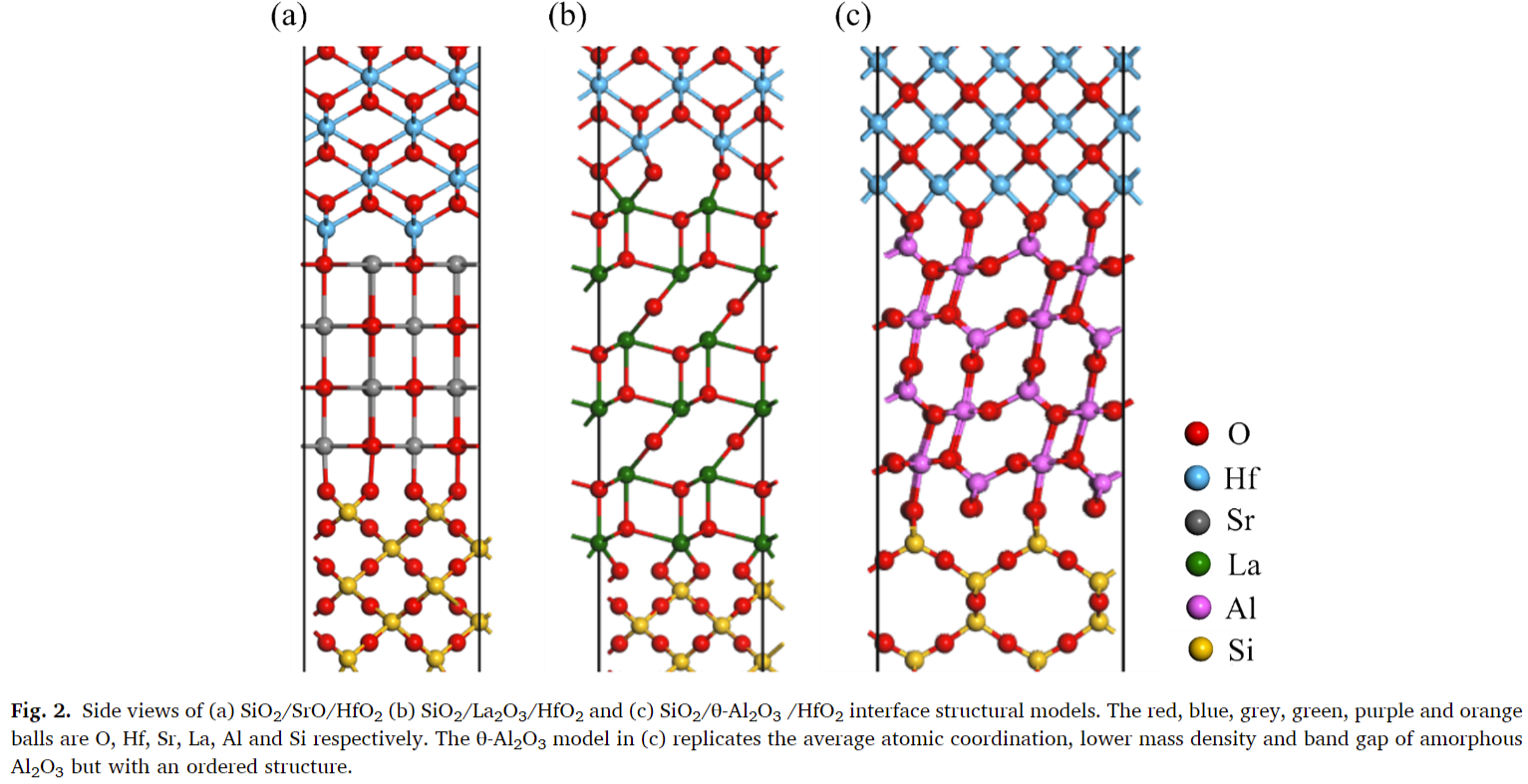
- 电子结构计算:直接计算了纯氧化物晶体薄片的电子结构,每个氧化物薄片由10层组成,以非极性(001)表面终止,并由15 Å真空层分隔。
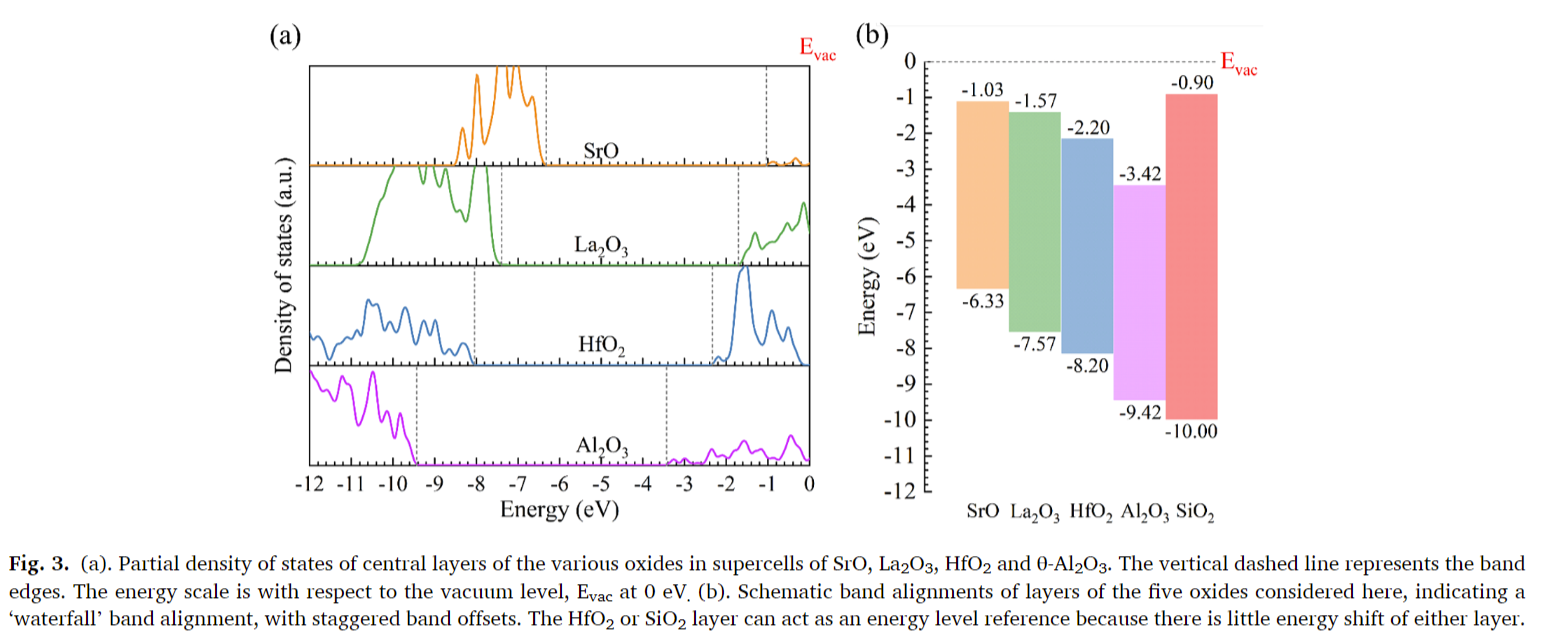
- 带隙和带对齐:氧化物的带隙和带对齐值与实验值或筛选交换计算值一致。
- 带对齐图:展示了四种氧化物的带对齐图,显示了由于它们相似的带隙,这些氧化物在化学顺序上与氧化物离子密度尺度给出的顺序相同。
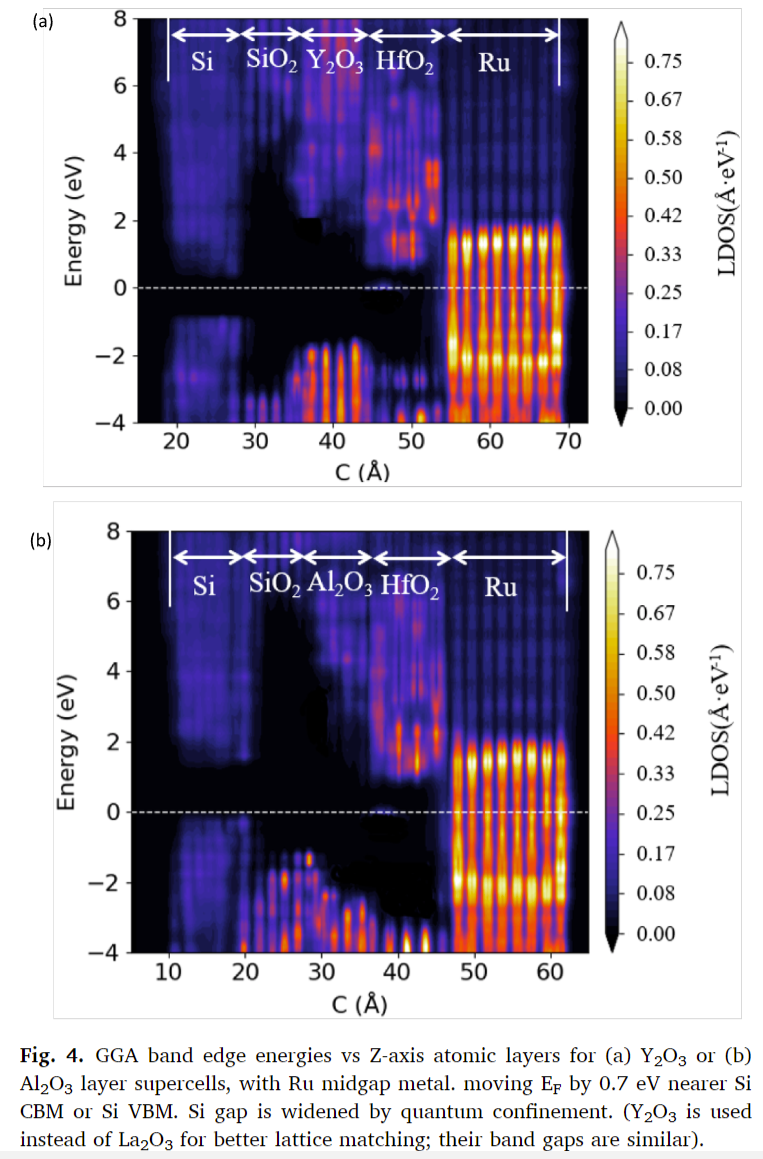
- 金属诱导的带隙态(MIGS):由于所有四种氧化物的电子介电常数ε∞较小,它们的S积分值较大,因此MIGS衰减较快,导致层间电子电荷转移很小,遵守电子亲和力规则。
4. 结论 (Conclusions)
文章得出结论,通过密度泛函理论分析,提出了一种电子结构模型,解释了金属/高K栅极堆叠中氧化物偶极工作函数偏移层的平坦带偏移的起源。平坦带电压偏移是由不同氧化物层之间的变化带对齐值引起的,这些值遵循交错的带对齐。不需要经验的离子性尺度。







![[Collection与数据结构] Map与Set(一):二叉搜索树与Map,Set的使用](https://img-blog.csdnimg.cn/direct/3069ea5b6d6347439610ad8cf557cd17.png)