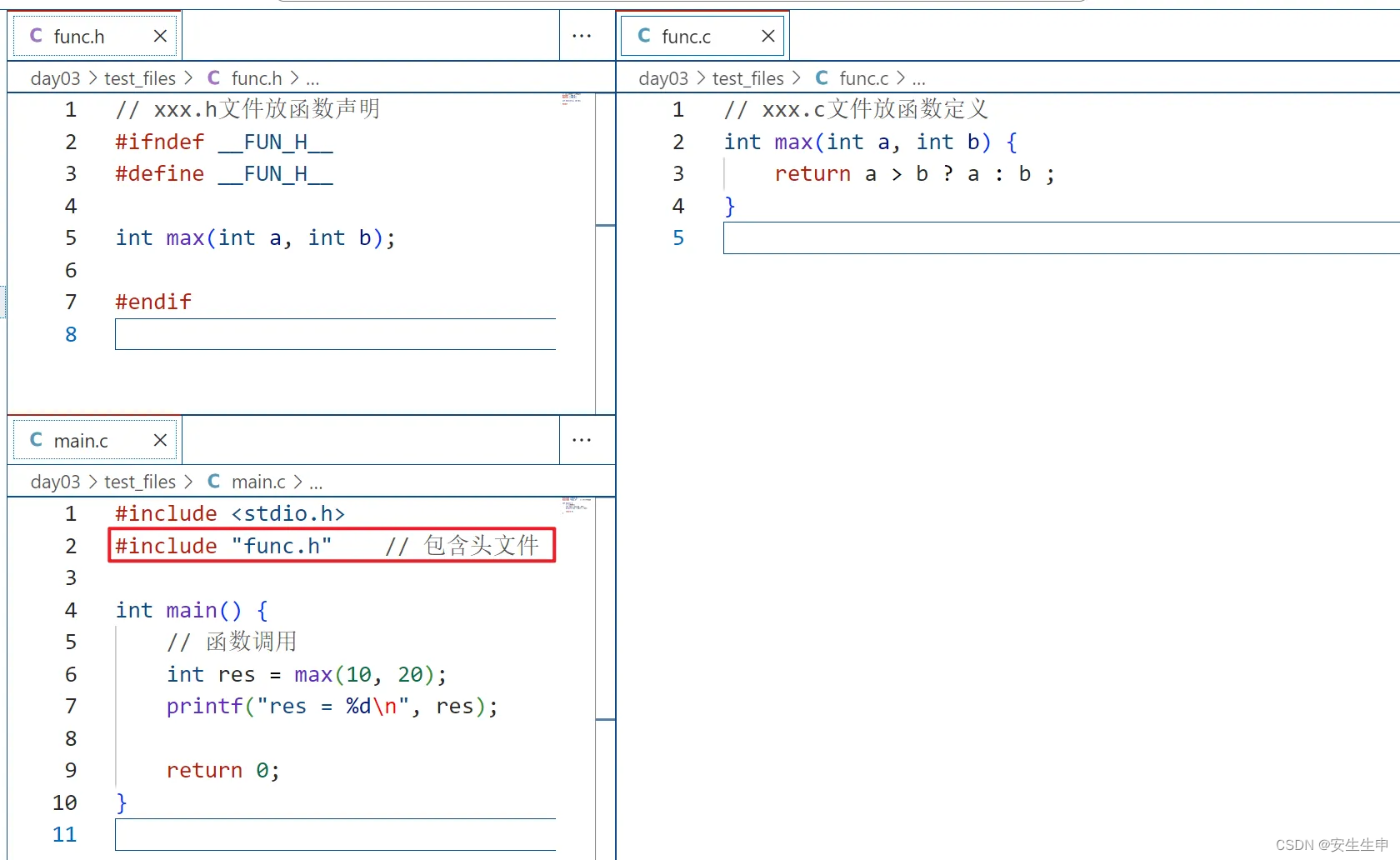
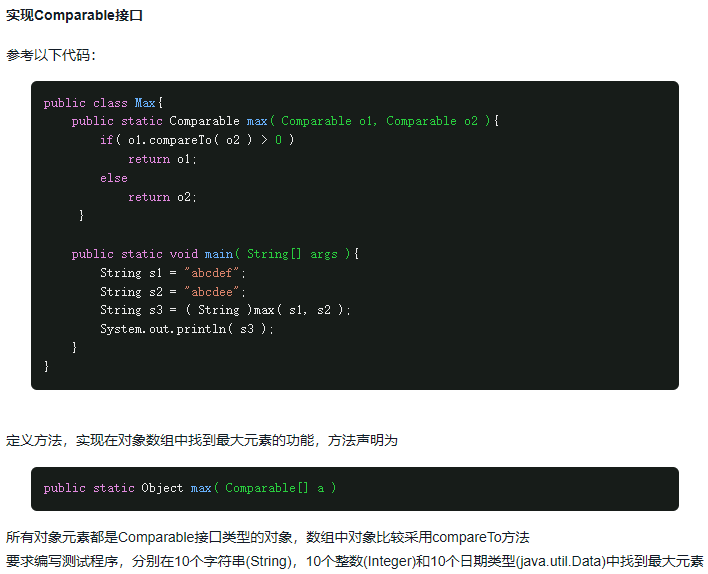
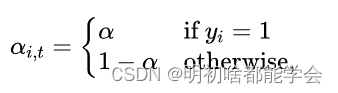本篇内容为scanpy的可视化方法,可以分为三部分:
- embedding的散点图;
- 用已知marker genes的聚类识别(Identification of clusters);
- 可视化基因的差异表达;
我们使用10x的PBMC数据集(包含68k个细胞)。Scanpy在其发行版中包含了这个数据集的缩减版,该数据集只包含700个细胞和765个高变基因。此数据集已经过预处理和UMAP计算。
在本篇内容里,我们使用到以下标记基因(来自于已知的文献结论,比如B-cell的标记基因为CD79A, MS4A1):
- B-cell: CD79A, MS4A1
- Plasma: IGJ (JCHAIN)
- T-cell: CD3D
- NK: GNLY, NKG7
- Myeloid: CST3, LYZ
- Monocytes: FCGR3A
- Dendritic: FCER1A
1. 降维的散点图(二维)
基于scanpy,tSNE、UMAP和其他几个embedding的散点图可以从文档轻松找到。比如可以看这里的选项列表:sc.pl.tsne,sc.pl.umap。
我们先进行初始化设置:
import scanpy as sc
import pandas as pd
from matplotlib.pyplot import rc_context
sc.set_figure_params(dpi=100, color_map = 'viridis_r')
sc.settings.verbosity = 1
sc.logging.print_header()
"""
scanpy==1.6.0 anndata==0.8.0 numpy==1.21.6 scipy==1.8.0 pandas==1.4.2 scikit-learn==1.0.2 statsmodels==0.11.0 python-igraph==0.8.0
"""
加载pbmc缩减版数据集:
pbmc = sc.datasets.pbmc68k_reduced()
# 检查pbmc内容
pbmc
"""
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
这里补充关于adata的obsm,varm和obsp的内容:
- obsm:对于观测的多维注释(即对于矩阵的行的多维注释),它是可变的ndarray,长度为n_obs,维度为2至多维。这里的m指的就是multi-dim多个维度的,obs_m对应于obs,但obs的每个成员都是一维的观测注释,obs_m的每个成员(X_pac和X_umap)都是多维的观测注释。
- varm:用于描述特征的,与obsm相对应。
- obsp(obs pair):针对观测的配对的注释(存储为稀疏矩阵),稀疏矩阵两维都是n_obs,obsp通常用于描述观测与观测之间的距离和连通性。比如:
pbmc.obsp['distances']
"""
<700x700 sparse matrix of type '<class 'numpy.float64'>'
with 6300 stored elements in Compressed Sparse Row format>
"""
行和列索引 距离
(0, 9) 8.365935325622559
(0, 54) 8.560888290405273
(0, 94) 7.486799716949463
......
(699, 695) 3.6524178981781006
1.1 可视化基因表达量
对于散点图,参数color是可视化的一个值,可以是Adata的任何基因或者obs里的任何对象,注意obs是存储注释信息的dataframe。
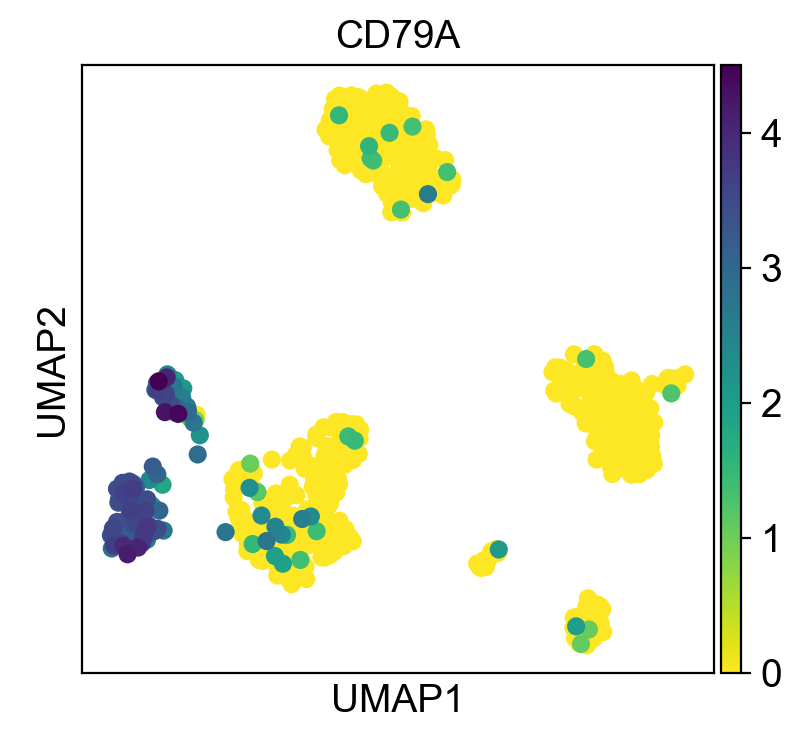
可视化基因CD79A在所有细胞中的表达量分布,由于pbmc这个adata已经有X_umap,我们可以用sc.pl.umap实现UMAP下的基因表达分布:
# rc_context用于指定figure大小
with rc_context({'figure.figsize': (4, 4)}):
sc.pl.umap(pbmc, color='CD79A')
可以给多个值。在下面的示例中,我们将绘制6个基因:“CD79A”、“MS4A1”、“IGJ”、“CD3D”、“FCER1A”和“FCGR3A”,以了解这些marker基因的表达。
此外,我们还将绘制另外两个值:
- 一个是每个细胞的UMI计数数
n_counts(UMI检测到的基因越多,数据越复杂); - 一个是一个分类值categorical value
bulk_labels(来自10x的细胞原始标签)。
使用参数ncols控制每行的可视化案例数。可以使用vmax调整绘制颜色的最大值(同样vmin可以用于最小值)。在本例中,我们使用vmax='p99',这意味着使用99%作为最大值。如果要分别为多个可视化案例设置vmax,则vamx可以是一个数字或一组数字。
此外,我们还使用frameon=False移除可视化图的边框,并用s=50设置点大小。
ncols = 4 表示画图一行4个图。
with rc_context({'figure.figsize': (3, 3)}):
sc.pl.umap(pbmc, color=['CD79A', 'MS4A1', 'IGJ', 'CD3D', 'FCER1A', 'FCGR3A', 'n_counts', 'bulk_labels'],
s=50, frameon=False, ncols=4, vmax='p99')
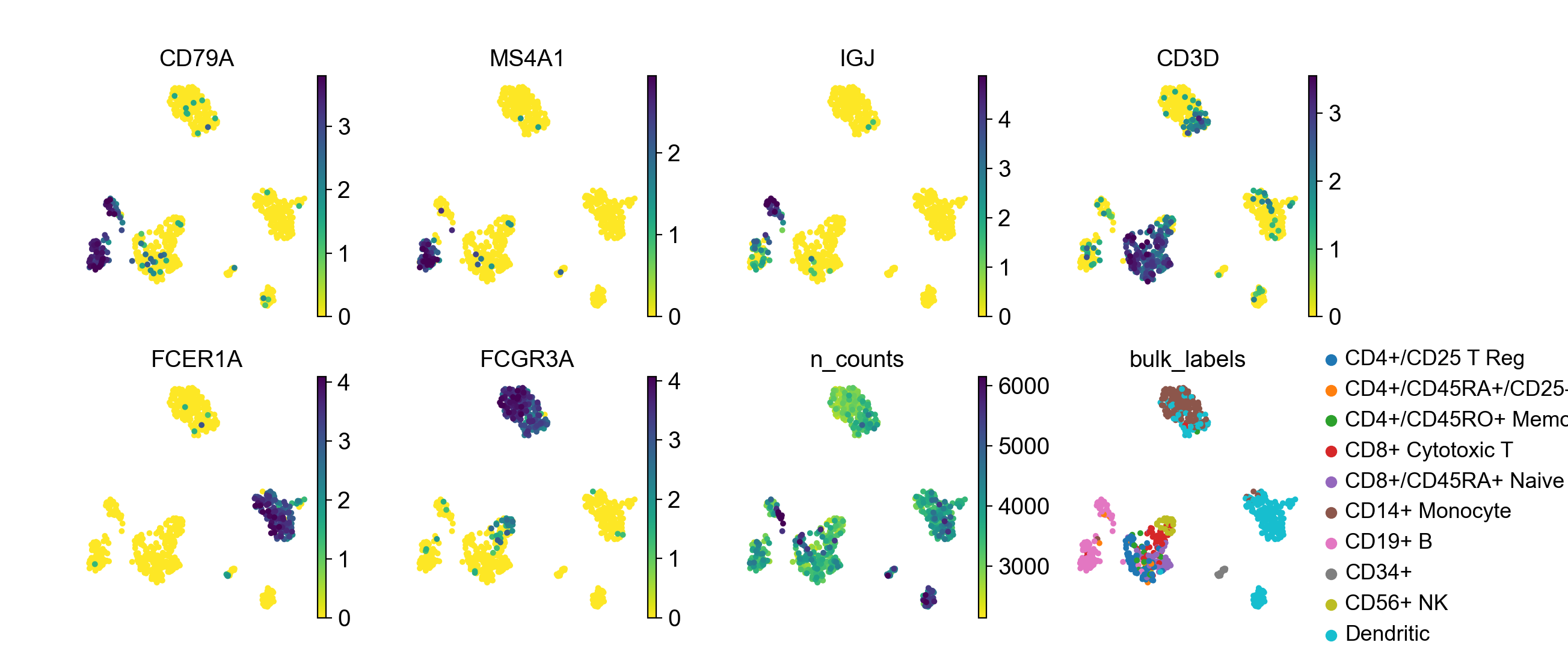
在图中,我们可以看到标记基因的细胞群与原始细胞标签的大概一致性。
散点图函数还有很多选项,可以微调图像。例如,我们可以如下所示查看clustering:
# 用leiden聚类计算, 结果存储到'clusters'中
sc.tl.leiden(pbmc, key_added='clusters', resolution=0.5)
pbmc
"""
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain', 'clusters'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups', 'leiden'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
注意到,obs中多了一个注释"clusters",下一步用"clusters"作为可视化的绘制值:
with rc_context({'figure.figsize': (5, 5)}):
sc.pl.umap(pbmc, color='clusters', add_outline=True, legend_loc='on data',
legend_fontsize=12, legend_fontoutline=2,frameon=False,
title='clustering of cells', palette='Set1')
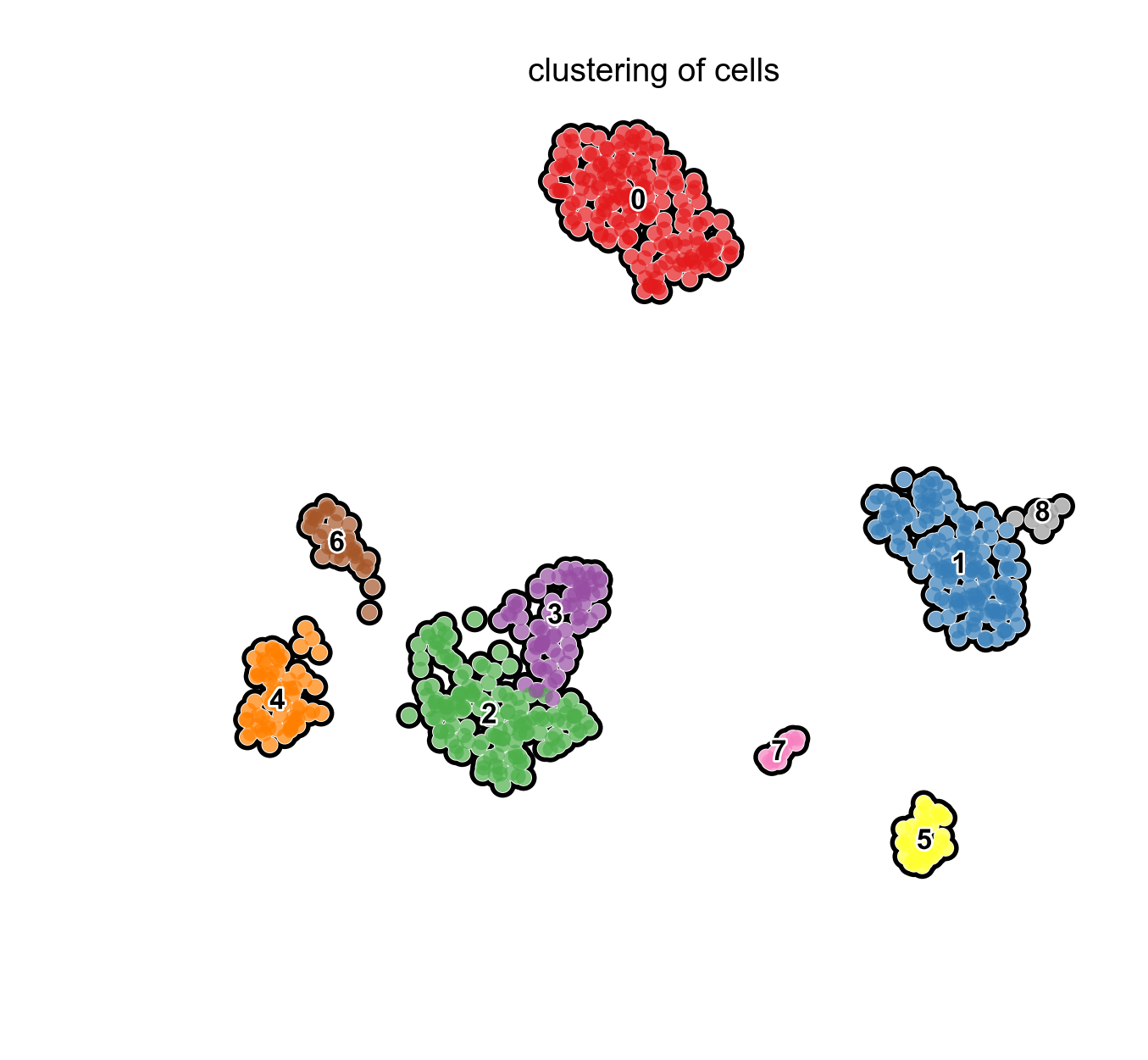
pbmc: 这是包含单细胞RNA测序数据的对象,可能是 Scanpy 中的AnnData对象。color='clusters': 这个参数指定了要在 UMAP 图上着色的变量,这里是细胞的聚类信息。add_outline=True: 这个参数指定是否为每个聚类添加轮廓线。legend_loc='on data': 这个参数指定图例的位置,‘on data’ 表示图例位于数据点的位置。legend_fontsize=12: 这个参数指定图例的字体大小。legend_fontoutline=2: 这个参数指定图例的字体轮廓线宽度。frameon=False: 这个参数指定是否显示图形周围的框架。title='clustering of cells': 这个参数指定图形的标题。palette='Set1': 这个参数指定了要使用的颜色调色板。‘
2. marker基因注释细胞类型
用已知marker基因识别clusters
通常,clusters需要使用已知的标记基因进行标记。使用散点图,我们可以看到一个基因在所有细胞中的表达,并可能将其与一个簇相关联。在这里,我们将展示使用点图dotplots、小提琴图violin plots、热图heatmaps和我们称之为“tracksplot”的图将标记基因与clusters关联。所有这些可视化都展示了相同的信息,最佳结果的选择由研究人员决定。
首先,我们为标记基因建立了一个字典,因为这将允许scanpy自动标记基因组:
marker_genes_dict = {
'B-cell': ['CD79A', 'MS4A1'],
'Dendritic': ['FCER1A', 'CST3'],
'Monocytes': ['FCGR3A'],
'NK': ['GNLY', 'NKG7'],
'Other': ['IGLL1'],
'Plasma': ['IGJ'],
'T-cell': ['CD3D'],
}
2.1 dotplot(点图)
使用 标记基因(marker_genes_dict)绘制成点图,进行细胞注释
检查每个簇中这些基因表达的快速方法是使用dotplot。这种图概括了两种类型的信息:
- 颜色表示一个基因,在每个类别(每个簇)内的平均表达;
- 点大小表示表达基因的细胞类别中的细胞比例。
此外,将树状图添加到图中也很有用(点图右侧的树状结构),可以将类似的簇聚集在一起。scanpy使用簇之间的PCA分量的相关性实现层次聚类。
sc.pl.dotplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True)
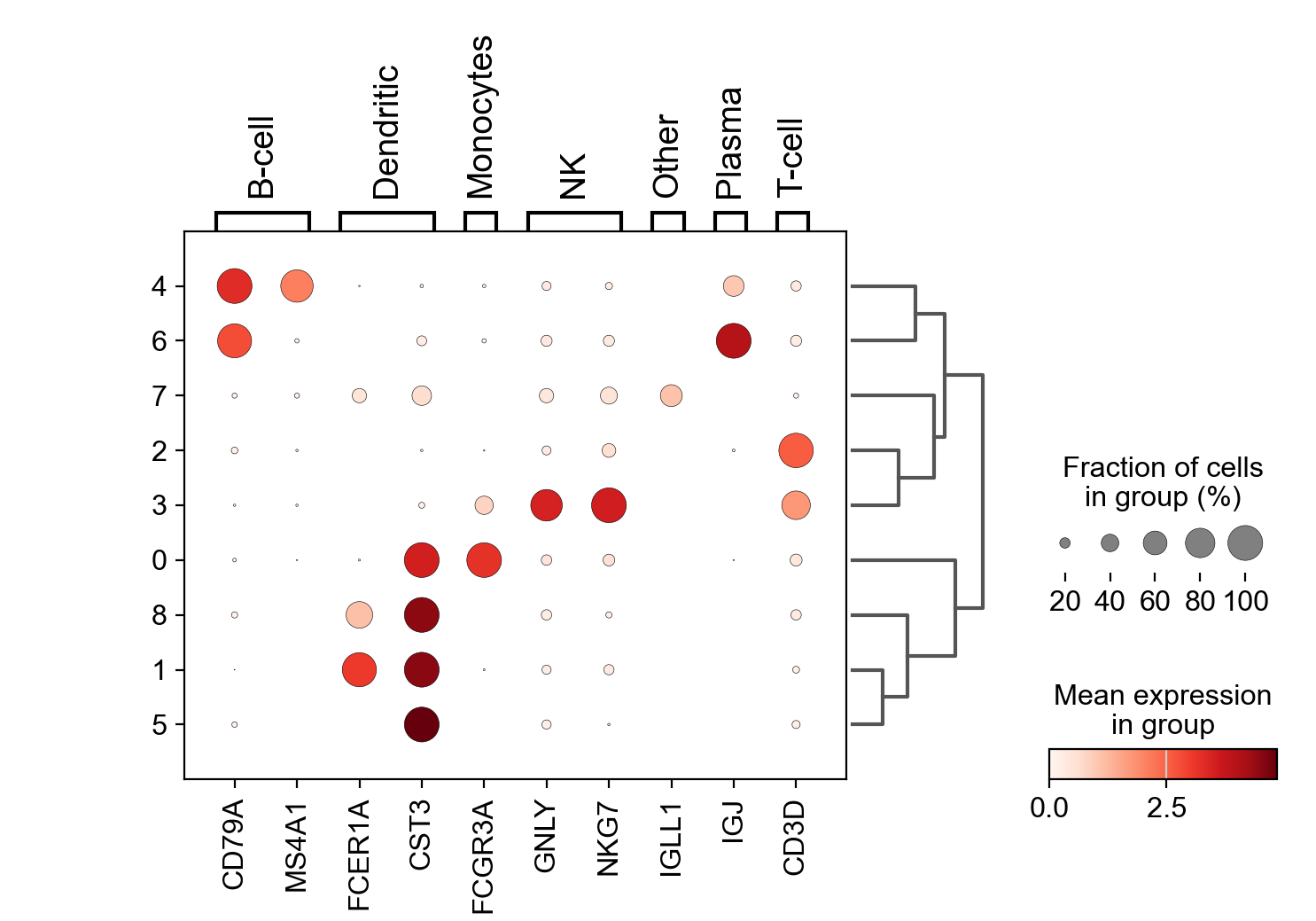 使用该图,我们可以看到簇4对应于B-cell,簇2对应于T-cell等。**该信息可用于手动注释细胞**,如下所示:
使用该图,我们可以看到簇4对应于B-cell,簇2对应于T-cell等。**该信息可用于手动注释细胞**,如下所示:
# 手动创建字典用于映射 簇 与 标签
cluster2annotation = {
'0': 'Monocytes',
'1': 'Dendritic',
'2': 'T-cell',
'3': 'NK',
'4': 'B-cell',
'5': 'Dendritic',
'6': 'Plasma',
'7': 'Other',
'8': 'Dendritic',
}
# 加入一个新的观测注释 `cell type` (使用pandas的map函数将clusters中的簇编号映射为cell type的标签)
pbmc.obs['cell type'] = pbmc.obs['clusters'].map(cluster2annotation).astype('category')
pbmc.obs['clusters']
"""
index
AAAGCCTGGCTAAC-1 0
AAATTCGATGCACA-1 0
AACACGTGGTCTTT-1 3
AAGTGCACGTGCTA-1 2
ACACGAACGGAGTG-1 1
..
TGGCACCTCCAACA-8 1
TGTGAGTGCTTTAC-8 0
TGTTACTGGCGATT-8 2
TTCAGTACCGGGAA-8 4
TTGAGGTGGAGAGC-8 1
Name: clusters, Length: 700, dtype: category
Categories (9, object): ['0', '1', '2', '3', ..., '5', '6', '7', '8']
"""
pbmc.obs['cell type']
"""
index
AAAGCCTGGCTAAC-1 Monocytes
AAATTCGATGCACA-1 Monocytes
AACACGTGGTCTTT-1 NK
AAGTGCACGTGCTA-1 T-cell
ACACGAACGGAGTG-1 Dendritic
...
TGGCACCTCCAACA-8 Dendritic
TGTGAGTGCTTTAC-8 Monocytes
TGTTACTGGCGATT-8 T-cell
TTCAGTACCGGGAA-8 B-cell
TTGAGGTGGAGAGC-8 Dendritic
Name: cell type, Length: 700, dtype: category
Categories (7, object): ['B-cell', 'Dendritic', 'Monocytes', 'NK', 'Other', 'Plasma', 'T-cell']
"""
我们用cell type去可视化点图:
# 使用 Scanpy 库将单细胞数据集(pbmc)上的标记基因(marker_genes_dict)绘制成点图,
# 并显示树形结构(dendrogram=True)。用`cell type`去可视化点图
sc.pl.dotplot(pbmc, marker_genes_dict, 'cell type', dendrogram=True)
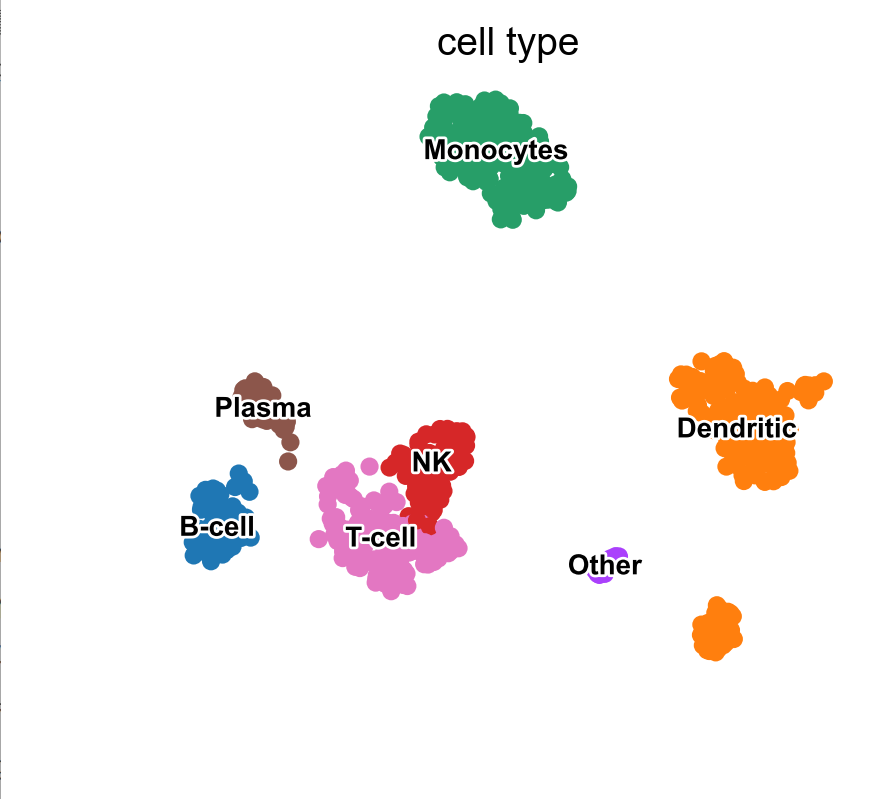
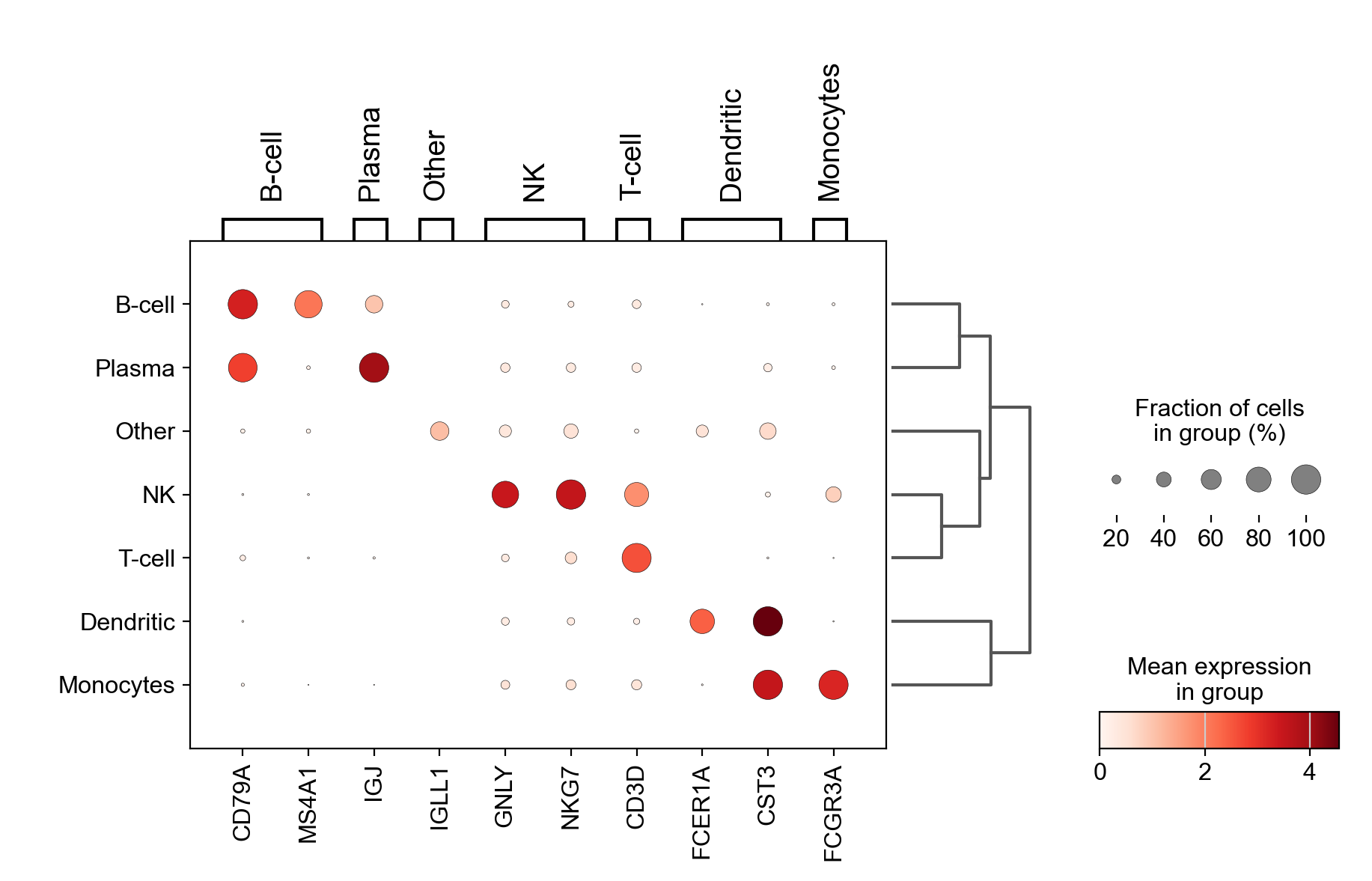 将标签可视化到**散点图**中,用`cell type`作为绘制的参数:
将标签可视化到**散点图**中,用`cell type`作为绘制的参数:
sc.pl.umap(pbmc, color='cell type', legend_loc='on data',
frameon=False, legend_fontsize=10, legend_fontoutline=2)
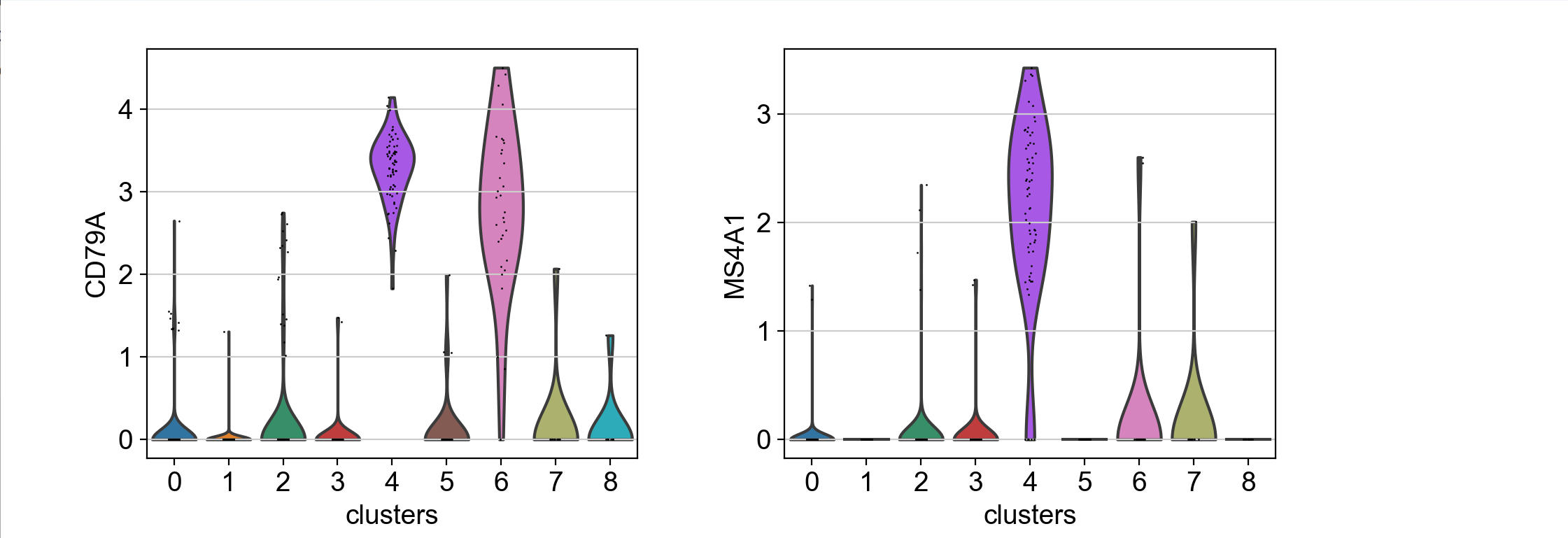
2.2 violin plot(小提琴图)
探索marker基因的另一种方式是violin plot。我们可以看到CD79A在簇4和簇6中的表达,以及MS4A1在簇4中的表达。(注意一个事实:CD79A和MS4A1是B-cell的标记基因)与dotplot相比,violin plot为我们提供了基因表达值在细胞中的分布概念。
使用clusters作为group的索引:
with rc_context({'figure.figsize': (4.5, 3)}):
sc.pl.violin(pbmc, ['CD79A', 'MS4A1'], groupby='clusters' )
小提琴图也可用于绘制存储在obs中的任何值。例如,这里使用小提琴图来比较不同簇之间的基因数量n_genes和线粒体基因百分比percent_mito。
with rc_context({'figure.figsize': (4.5, 3)}):
# stripplot=False用于删除internal dots, inner='box'用于在小提琴内部加方框图
sc.pl.violin(pbmc, ['n_genes', 'percent_mito'], groupby='clusters', stripplot=False, inner='box')
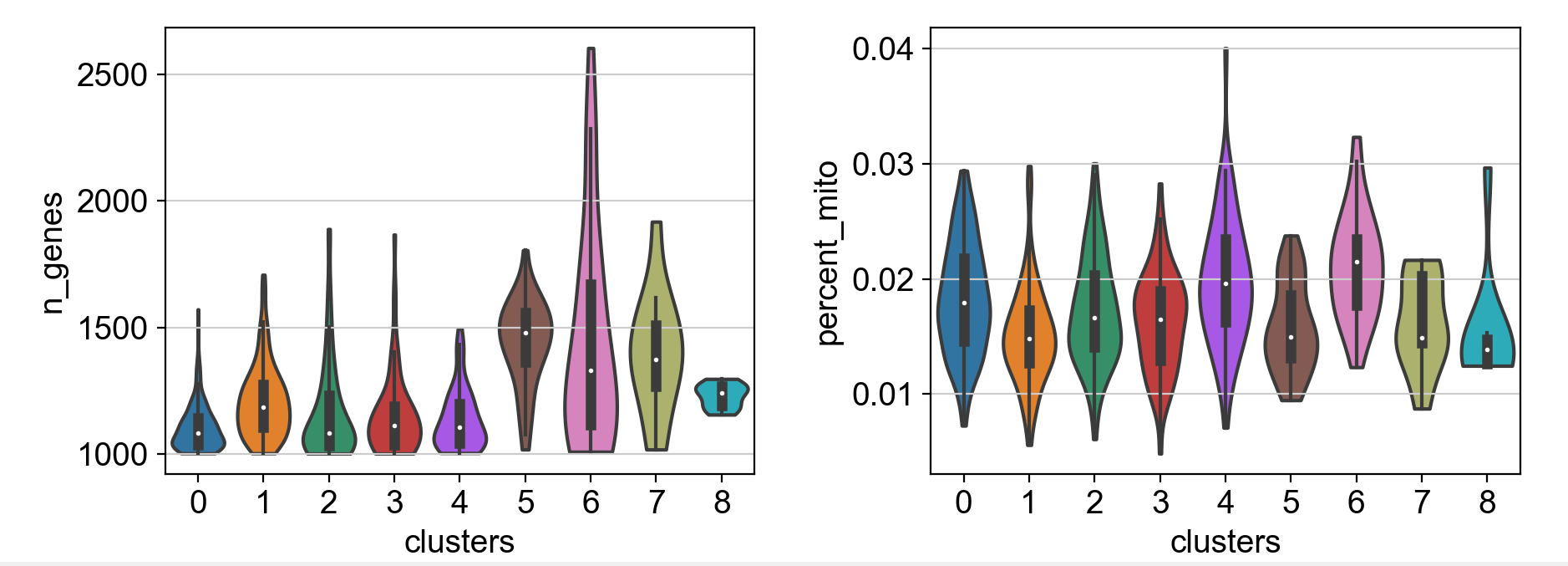
参数解释:
stripplot=False:默认情况下,小提琴图的内部会显示数据点的分布,有时候这些数据点可能会重叠在一起,使得图形不够清晰。将stripplot参数设置为False可以删除这些内部的数据点,只保留小提琴的形状,使得图形更清晰。
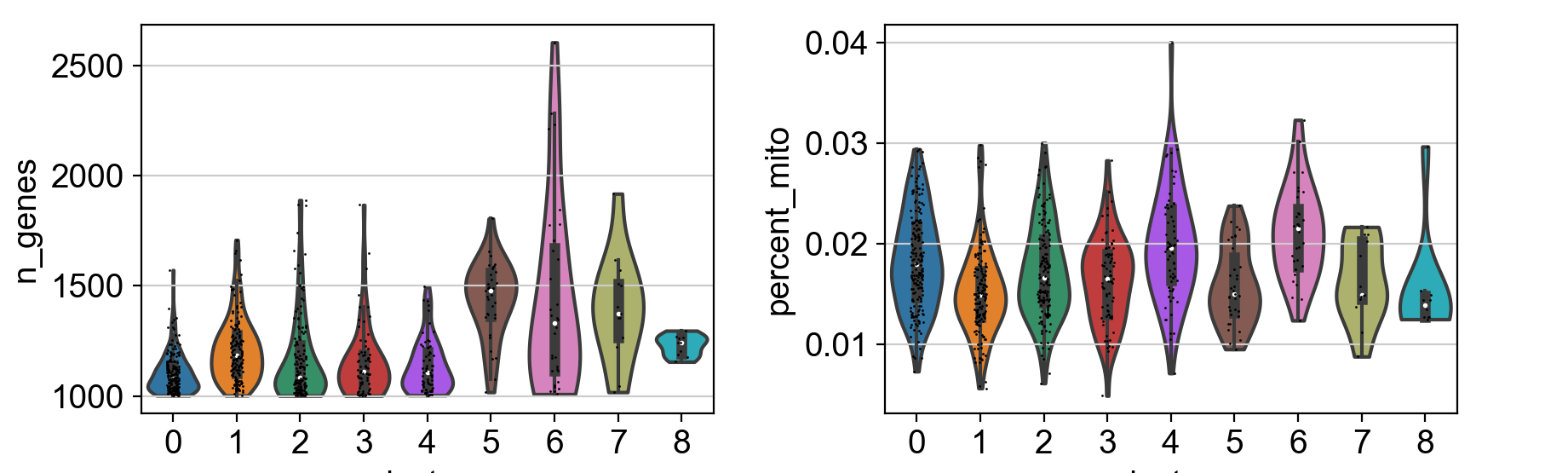
-
inner='box':小提琴图的内部形状表示了数据的分布情况。默认情况下,内部形状是一个核密度估计的曲线,显示了数据的分布密度。而设置inner='box'则会在小提琴内部添加一个箱线图(Box plot),用于显示数据的五数概括(最小值、第一四分位数、中位数、第三四分位数、最大值),这有助于更直观地理解数据的分布情况。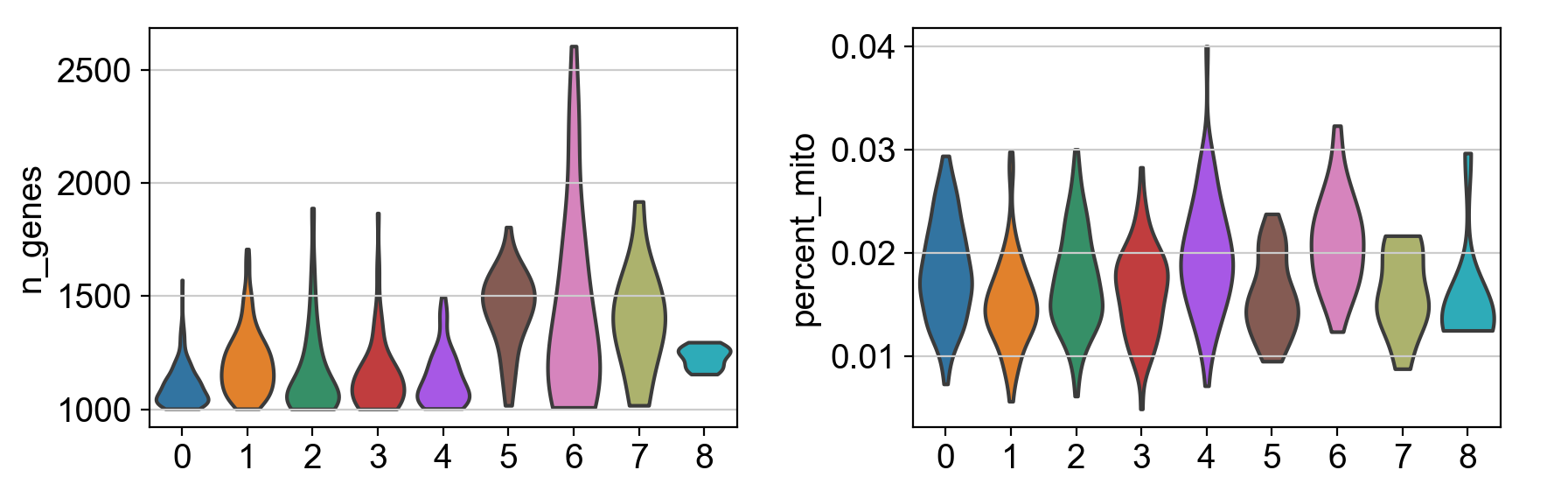
2.3 stacked-violin plot(all gene)
为了同时查看所有marker基因的小提琴图,我们使用sc.pl.stacked_violin,如前所述,添加了一个树状图(dendrogram)来对相似的簇进行分组。
ax = sc.pl.stacked_violin(pbmc, marker_genes_dict, groupby='clusters',
swap_axes=False, dendrogram=True)
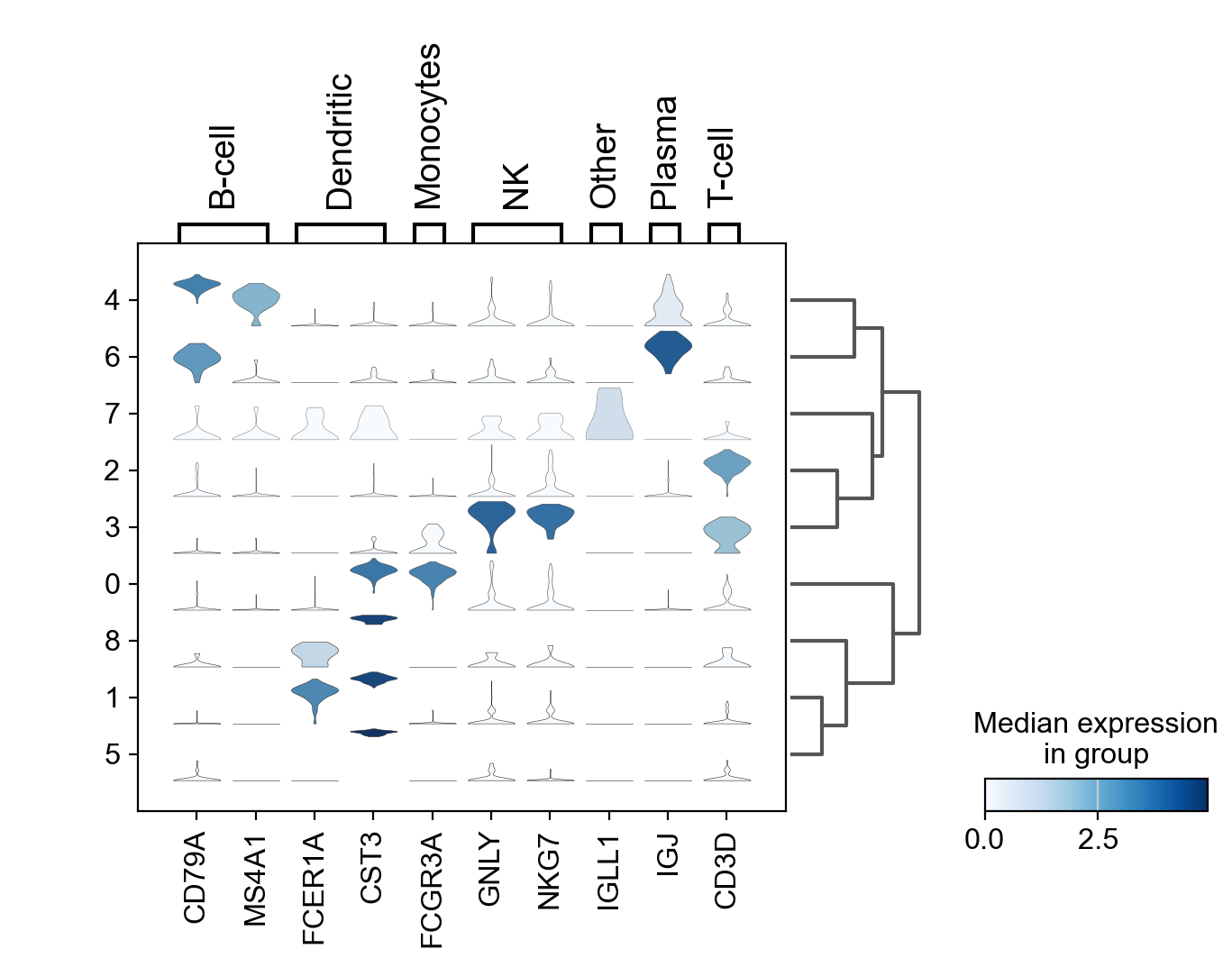
参数解释:
swap_axes=False:默认情况下,堆叠小提琴图的 x 轴是样本类别(例如不同的聚类群体),y 轴是基因的表达值。将swap_axes参数设置为False表示不交换 x 轴和 y 轴。如果设置为True,则会交换 x 轴和 y 轴,使得聚类群体显示在 y 轴上,基因表达值显示在 x 轴上。dendrogram=True:设置为True时,在堆叠小提琴图的左侧会显示一个聚类树状图(Dendrogram),用于展示基因的聚类关系。这个聚类树状图通常是通过聚类算法对基因进行聚类得到的,它显示了基因之间的相似性或相关性,有助于理解基因的分组结构。
2.4 matrixplot(矩阵图)
可视化基因表达的一种简单方法是使用matrixplot,这是按类别(簇)分组的每个基因的平均表达值的热图。这种图基本上显示与点图中的颜色相同的信息。
在这里,将基因的表达从0缩放到1,这是最大的平均表达,0是最小的。
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True, cmap='Blues',
standard_scale='var', colorbar_title='column scaled\nexpression')
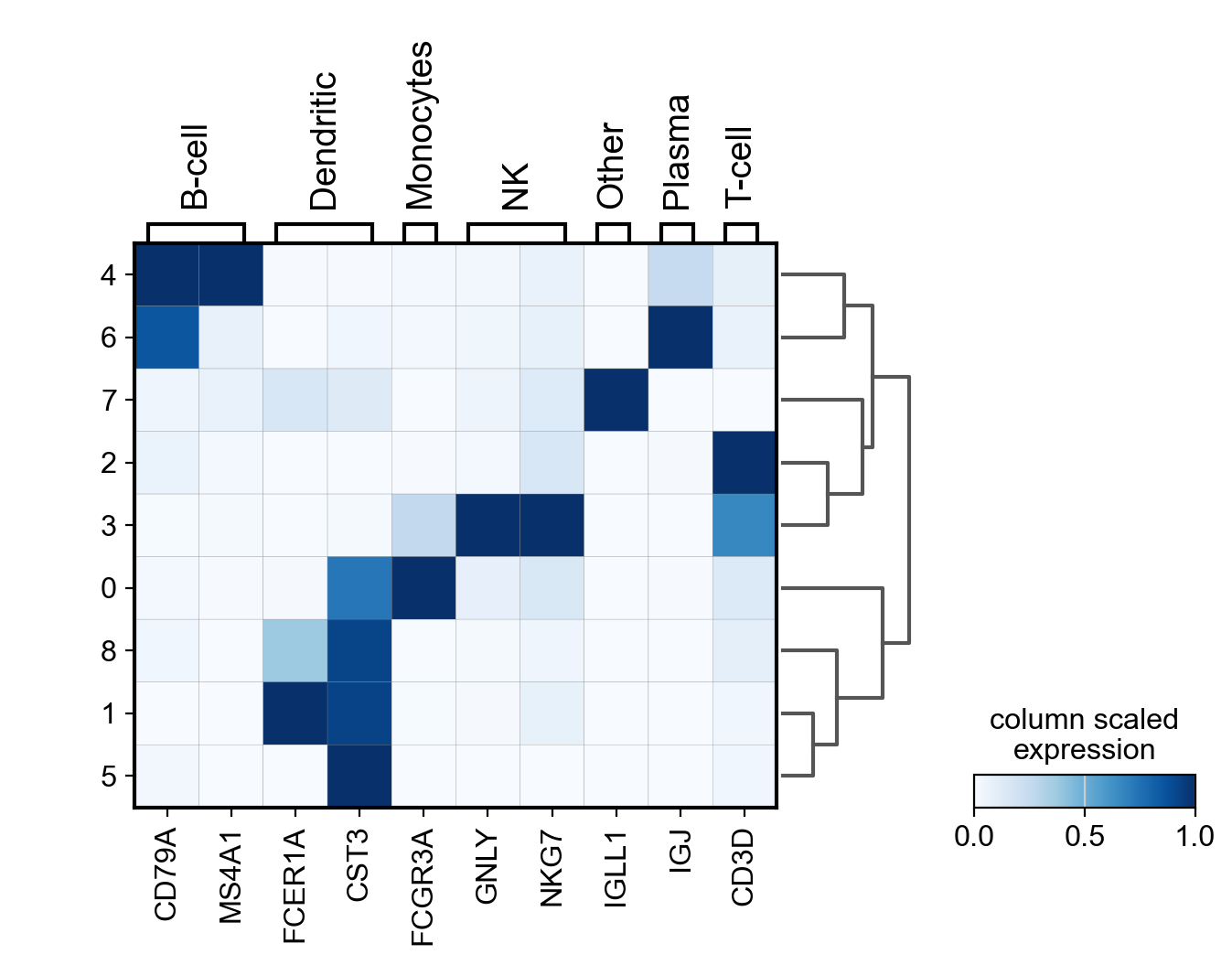
参数解释:
cmap='Blues':这个参数指定了矩阵图的颜色映射。在这里,'Blues'表示使用蓝色系列的颜色映射,即较低的值使用较浅的蓝色,较高的值使用较深的蓝色。standard_scale='var':这个参数控制是否对数据进行标准化。在这里,设置为'var'表示按列(即基因)进行方差标准化。colorbar_title='column scaled\nexpression':这个参数指定了颜色条的标题。在这里,标题被设置为'column scaled\nexpression',这意味着颜色条显示的是经过列(基因)标准化的表达量。\n表示换行符,因此标题会显示在两行上。
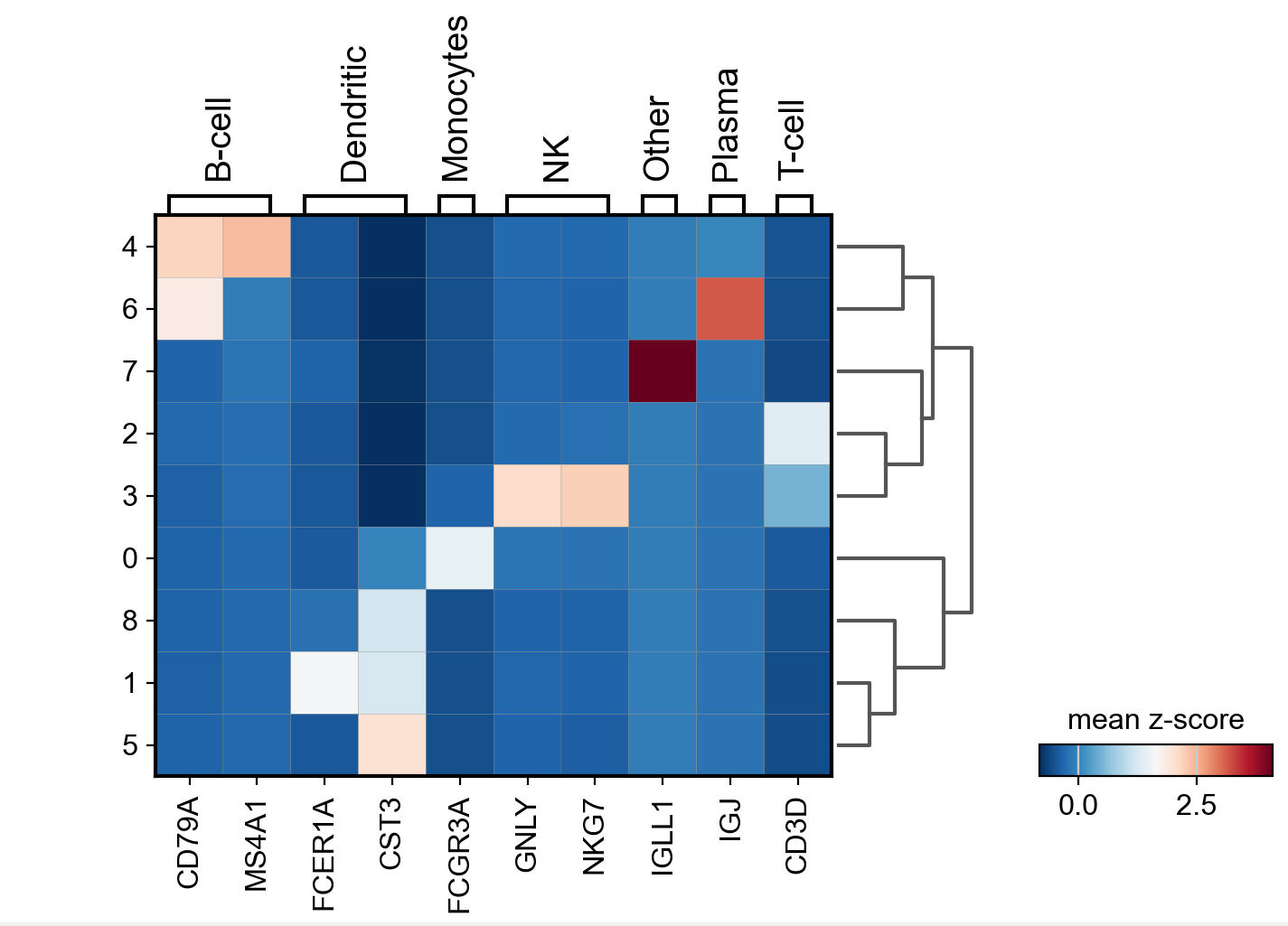
ax = sc.pl.matrixplot(pbmc, marker_genes_dict, groupby='clusters',
dendrogram=True, swap_axes=False)
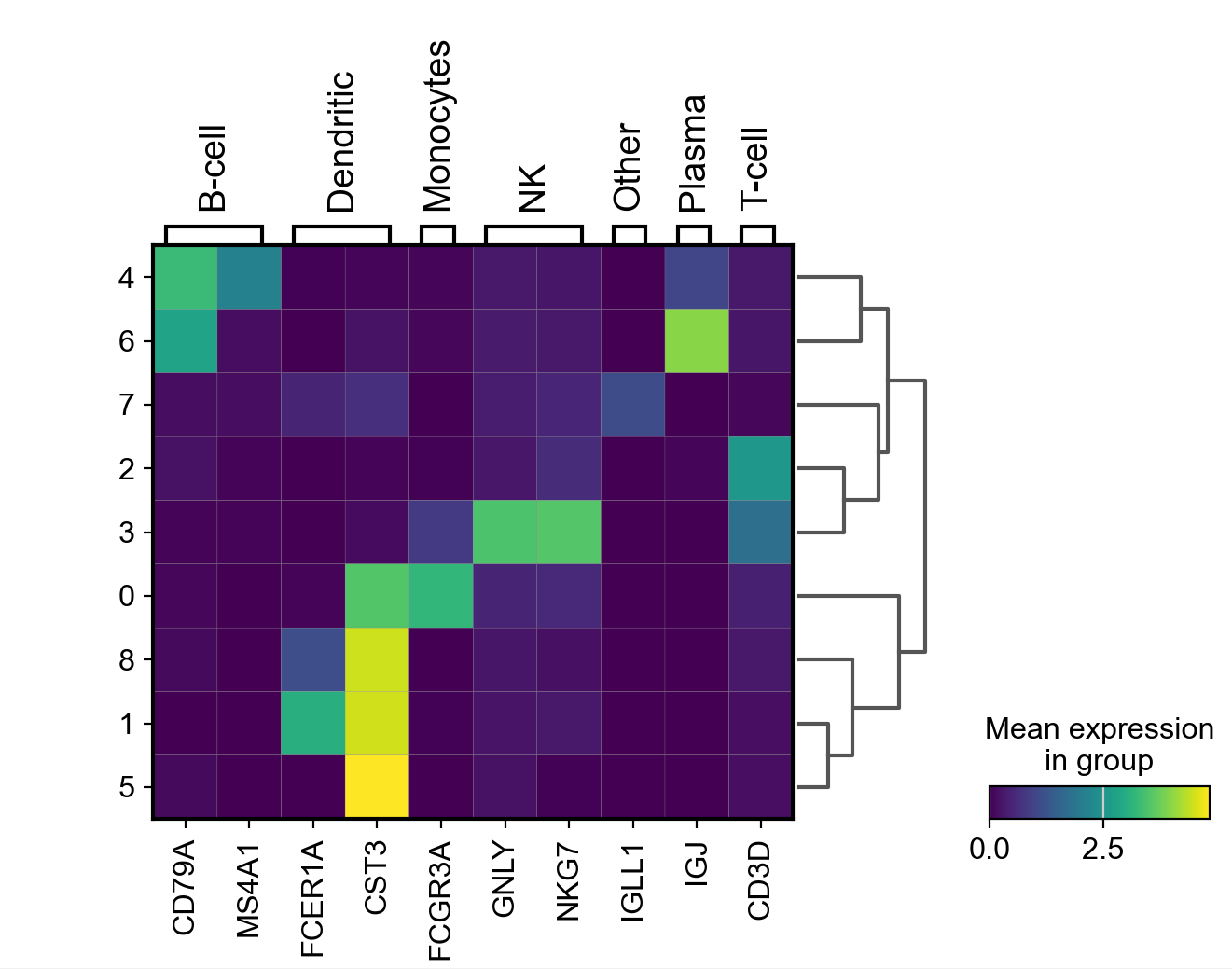 其他有用的选项是用`sc.pp.scale`对表达标准化,这里,我们将scale后的信息保存在adata的隐藏dataframe`layers`中:
其他有用的选项是用`sc.pp.scale`对表达标准化,这里,我们将scale后的信息保存在adata的隐藏dataframe`layers`中:
pbmc.layers['scaled'] = sc.pp.scale(pbmc, copy=True).X
print(pbmc)
'''
AnnData object with n_obs × n_vars = 700 × 765
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain', 'clusters', 'cell type'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups', 'leiden'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'scaled'
obsp: 'distances', 'connectivities'
'''
再用matrixplot可视化,并使用其他cmap:
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True,
colorbar_title='mean z-score', layer='scaled', cmap='RdBu_r')
参数解释:
cmap='RdBu_r'是指定了矩阵图的颜色映射。在这里,'RdBu_r'是一种颜色映射方案,具体含义如下:
'RdBu_r'是由三个部分组成的,分别是'Rd'、'Bu'和'r'。
'Rd'代表红色(Red)到白色(White)的渐变,表示正值。在这种颜色映射中,较小的值(即较低的表达量)会显示为红色,而较大的值(即较高的表达量)会显示为白色。
'Bu'代表蓝色(Blue)到白色(White)的渐变,表示负值。在这种颜色映射中,较小的值(即较低的表达量)会显示为蓝色,而较大的值(即较高的表达量)会显示为白色。
'r'表示颜色映射的反转,即从'Rd'开始到'Bu'结束。因此,
cmap='RdBu_r'意味着使用红色到白色和蓝色到白色的渐变来表示正负值,表达量越高的基因会显示为白色,表达量越低的基因会显示为红色或蓝色。
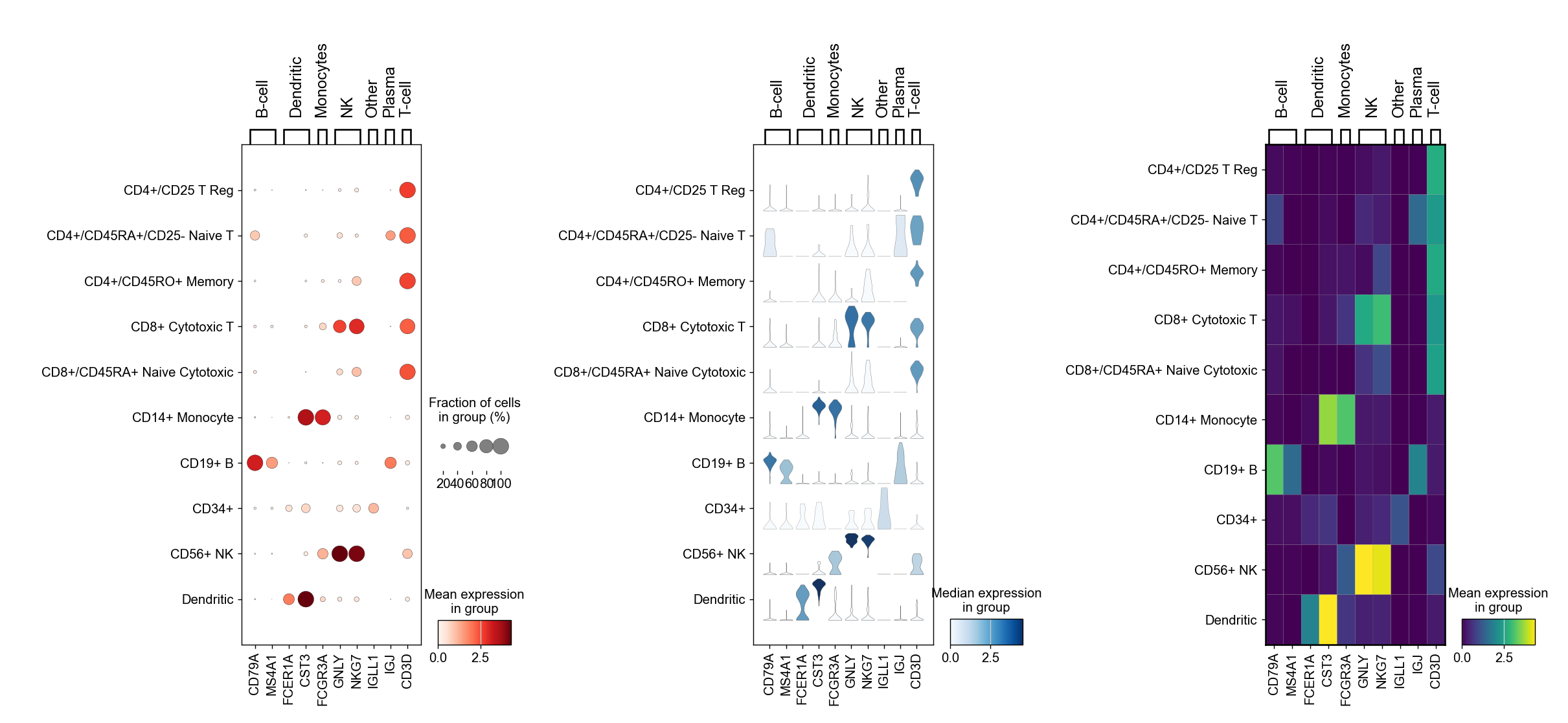
3. 集成多图到一张图展示_subplot
可以使用plt.subplots的返回对象ax将图变成子图:
import matplotlib.pyplot as plt
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(20,4), gridspec_kw={'wspace':0.9})
ax1_dict = sc.pl.dotplot(pbmc, marker_genes_dict, groupby='bulk_labels', ax=ax1, show=False)
ax2_dict = sc.pl.stacked_violin(pbmc, marker_genes_dict, groupby='bulk_labels', ax=ax2, show=False)
ax3_dict = sc.pl.matrixplot(pbmc, marker_genes_dict, groupby='bulk_labels', ax=ax3, show=False, cmap='viridis')
plt.show()
4. Heatmaps
热图不像前面的图会折叠了每个细胞,热图中,每个细胞都显示在每一行上。
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', cmap='viridis', dendrogram=True)
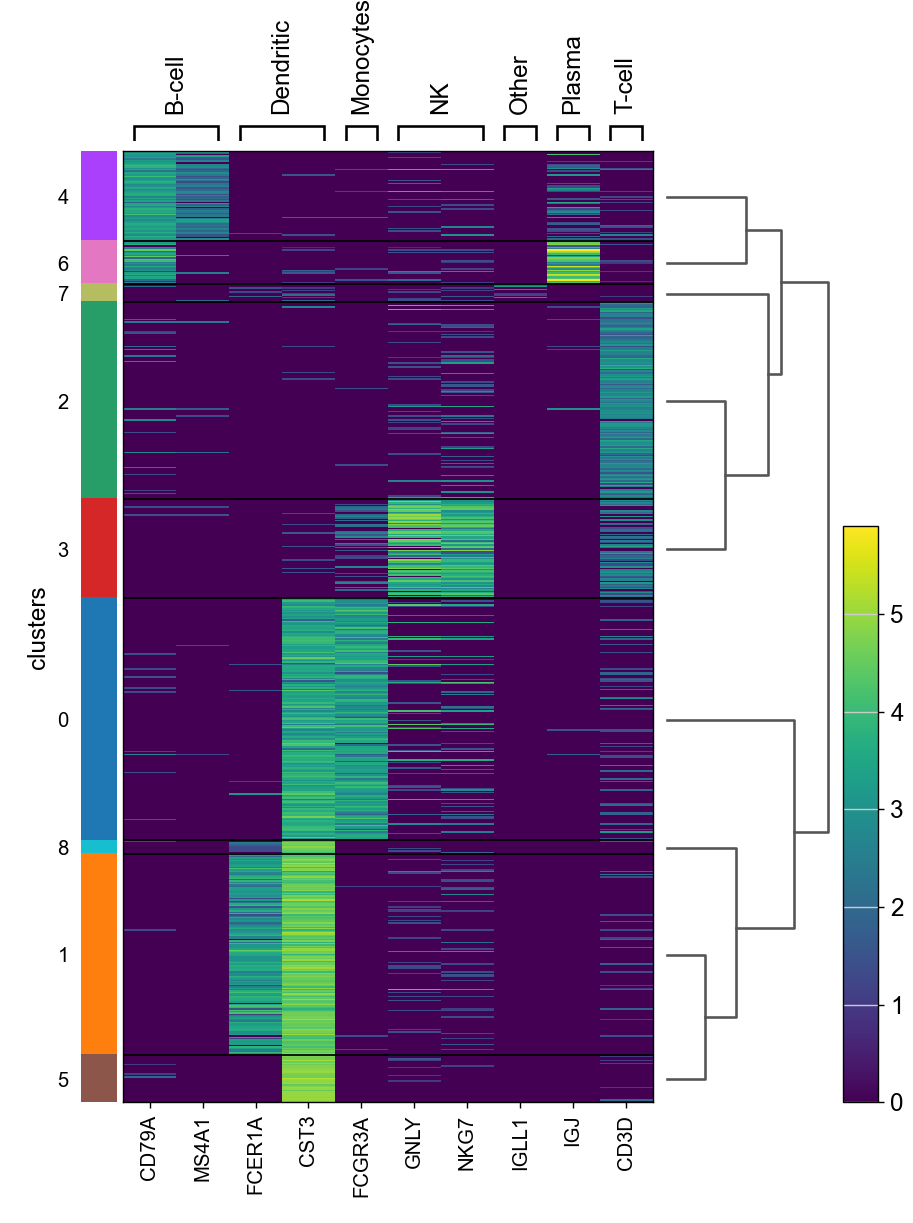 热图也可以绘制在scale过的数据上。在下图中,与前面的matrixplot相似(注意参数`layer='scaled`)。
热图也可以绘制在scale过的数据上。在下图中,与前面的matrixplot相似(注意参数`layer='scaled`)。
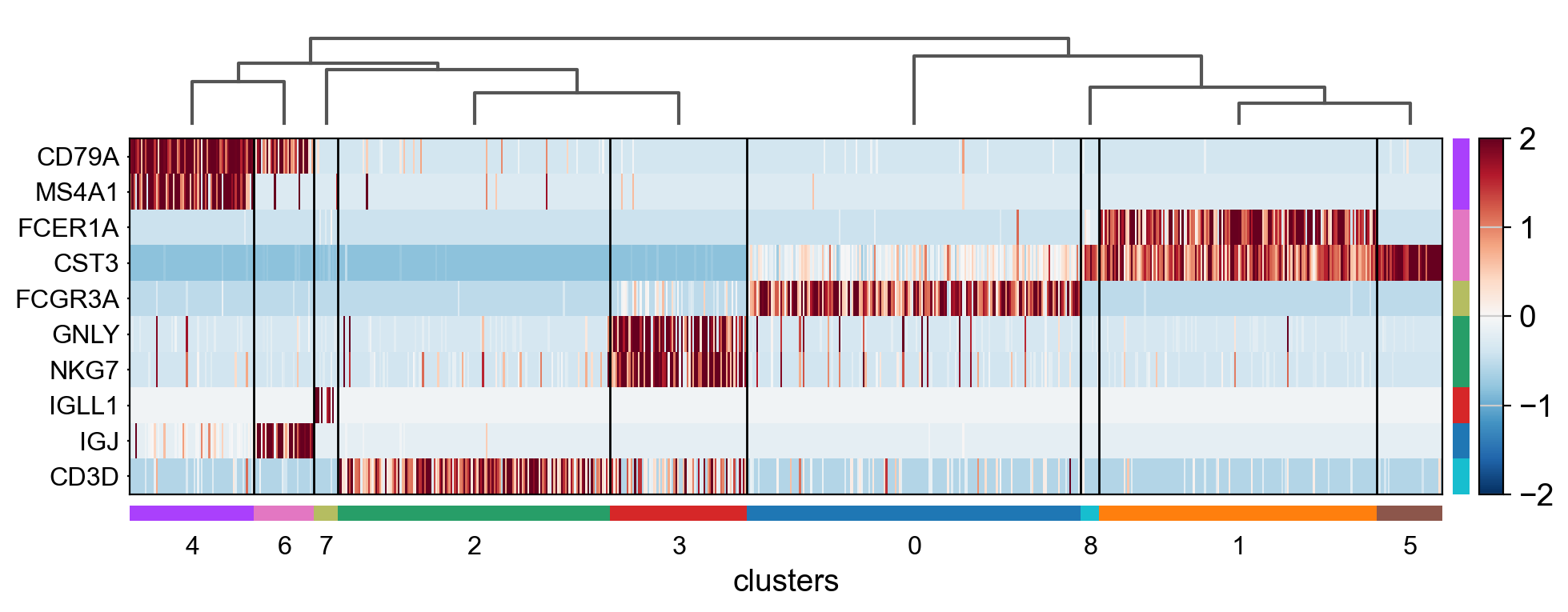
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', layer='scaled',
vmin=-2, vmax=2, cmap='RdBu_r', dendrogram=True, swap_axes=True, figsize=(11,4))
参数解释:
layer='scaled':这个参数指定了要绘制的数据层。在这里,'scaled'表示绘制经过缩放处理的数据,即标准化或归一化后的数据。vmin=-2, vmax=2:这两个参数分别指定了颜色映射的最小值和最大值。在这里,vmin=-2表示颜色映射的最小值为 -2,vmax=2表示颜色映射的最大值为 2。任何小于 -2 的值将显示为颜色映射的最小值,任何大于 2 的值将显示为颜色映射的最大值。cmap='RdBu_r':这个参数指定了热图的颜色映射。在这里,'RdBu_r'是一种颜色映射方案,它从深蓝色到深红色渐变,用于表示数值的正负变化。因此,较小的值会显示为深蓝色,而较大的值会显示为深红色。dendrogram=True:设置为True时,在热图的顶部和左侧会显示聚类树状图(Dendrogram),用于展示基因或细胞的聚类关系。swap_axes=True:设置为True时,将交换热图的 x 轴和 y 轴。换句话说,细胞或样本将显示在热图的横轴上,而基因或特征将显示在热图的纵轴上。figsize=(11,4):这个参数指定了绘制热图的图形大小。在这里,热图的宽度为 11 英寸,高度为 4 英寸。
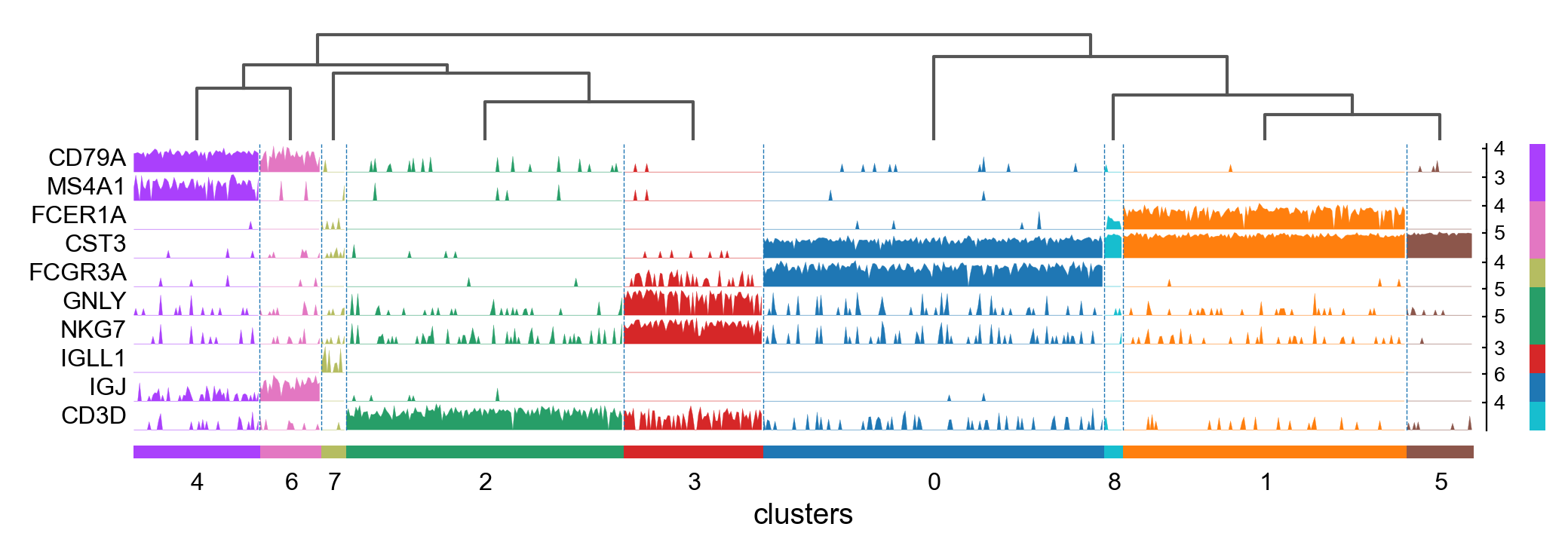
5. Tracksplot(轨迹图)
绘制轨迹图
轨迹图(Tracks Plot)是一种可视化方法,用于显示单细胞数据中基因或特征在不同细胞群体之间的表达差异。通过将不同的基因或特征绘制成轨迹,可以直观地观察到它们在细胞群体之间的表达模式的差异和相似性。
Tracksplot显示的信息与热图相同,但基因表达由高度表示,而不是色阶。
ax = sc.pl.tracksplot(pbmc, marker_genes_dict, groupby='clusters', dendrogram=True)
6. 关键基因可视化
我们可以识别在簇中差异表达的基因,而不是像前面那样通过已知的标记基因来表示簇。
我们用sc.tl.rank_genes_groups识别差异表达的基因,此函数将获取每组细胞,并将每组中每个基因的分布与不在该组中的所有其他细胞中的分布进行比较。
sc.tl.rank_genes_groups(pbmc, groupby='clusters', method='wilcoxon')
在这里,我们将使用10x给出的原始细胞标签来验证这些细胞类型的标记基因。
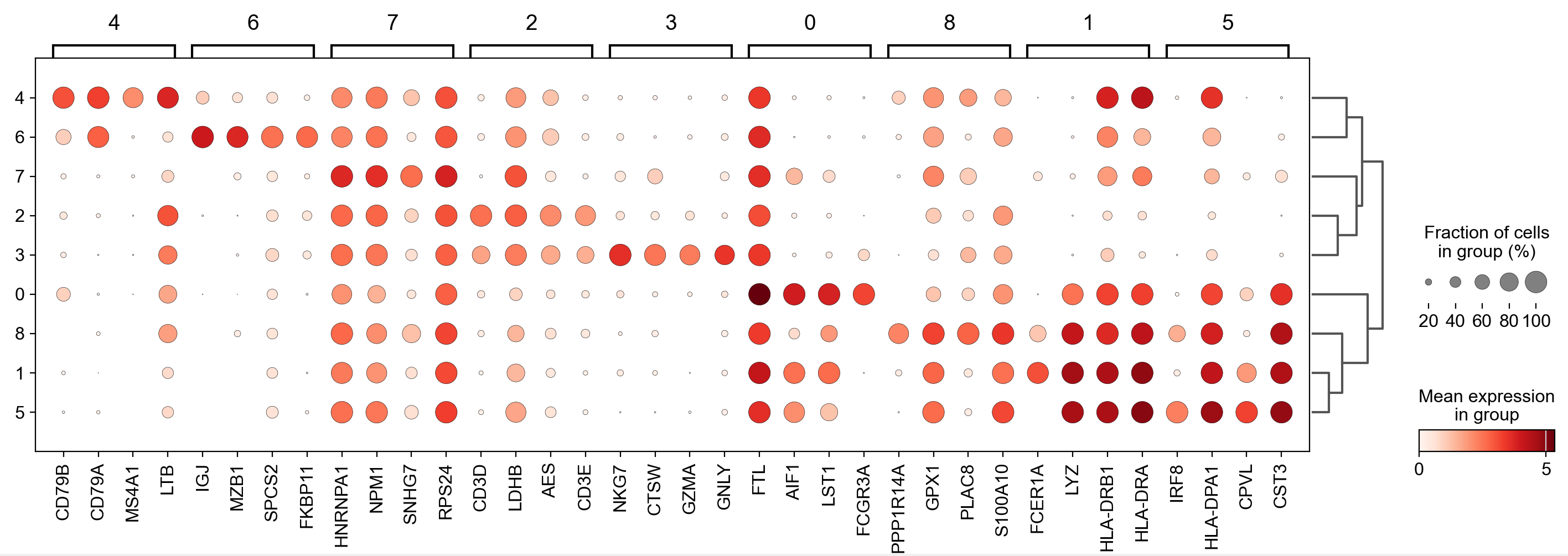
6.1 dotplot可视化marker基因
dotplot可以得到大致的差异基因表达总览。为了让可视化结果更紧凑,我们仅对每类细胞的差异表达得分前4的基因可视化(n_genes=4)。
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4)
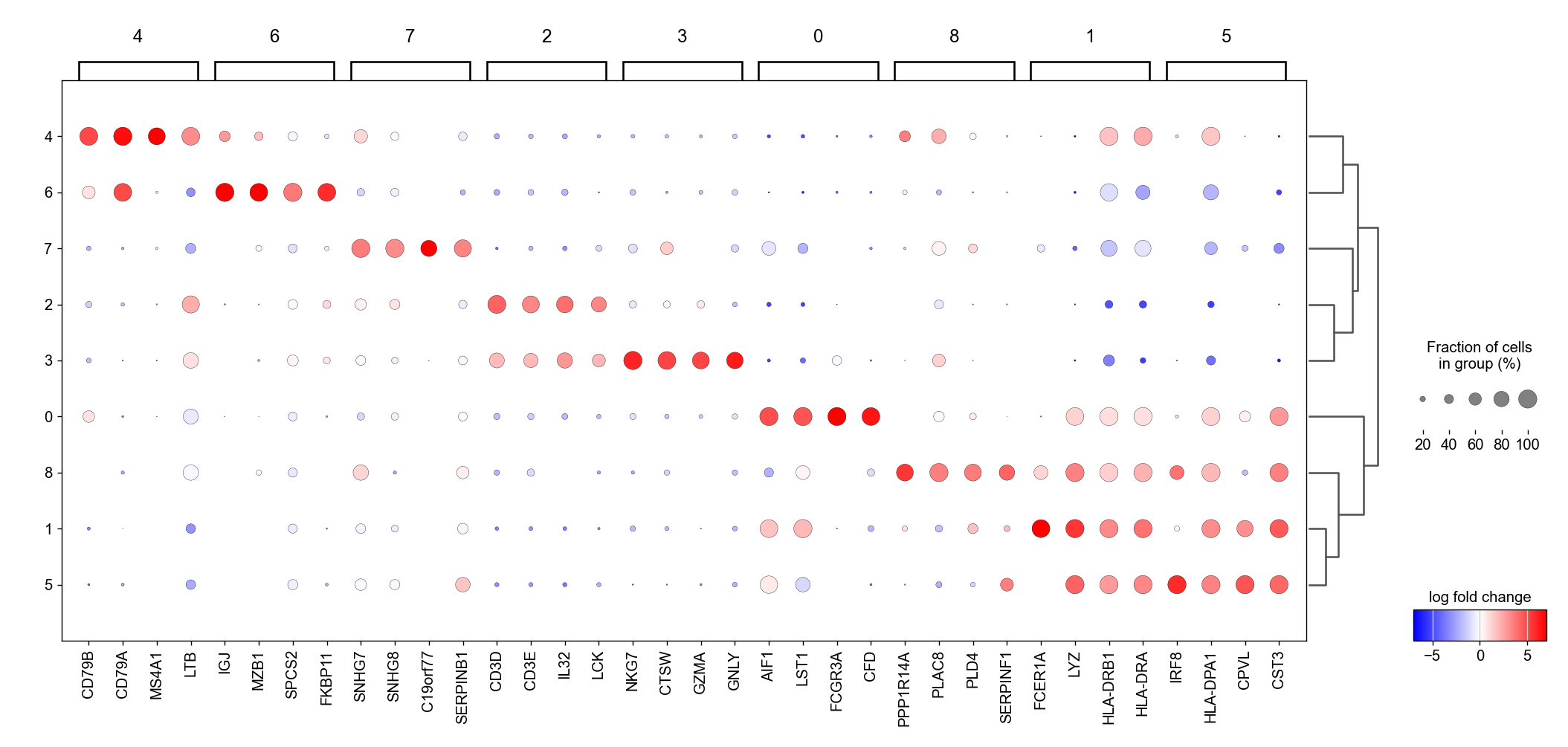
为了获得更好的表示,我们可以可视化取log(values_to_plot='logfoldchanges'),而不是原始的基因表达图。此外,我们希望关注差异得分取log后>=3的基因(min_logfoldchange=3)。
由于log是一个发散比例,我们还要调整可视化的最小值和最大值。注意,在下面的图中,很难区分T-cell。
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4, values_to_plot='logfoldchanges', min_logfoldchange=3,
vmax=7, vmin=-7, cmap='bwr')
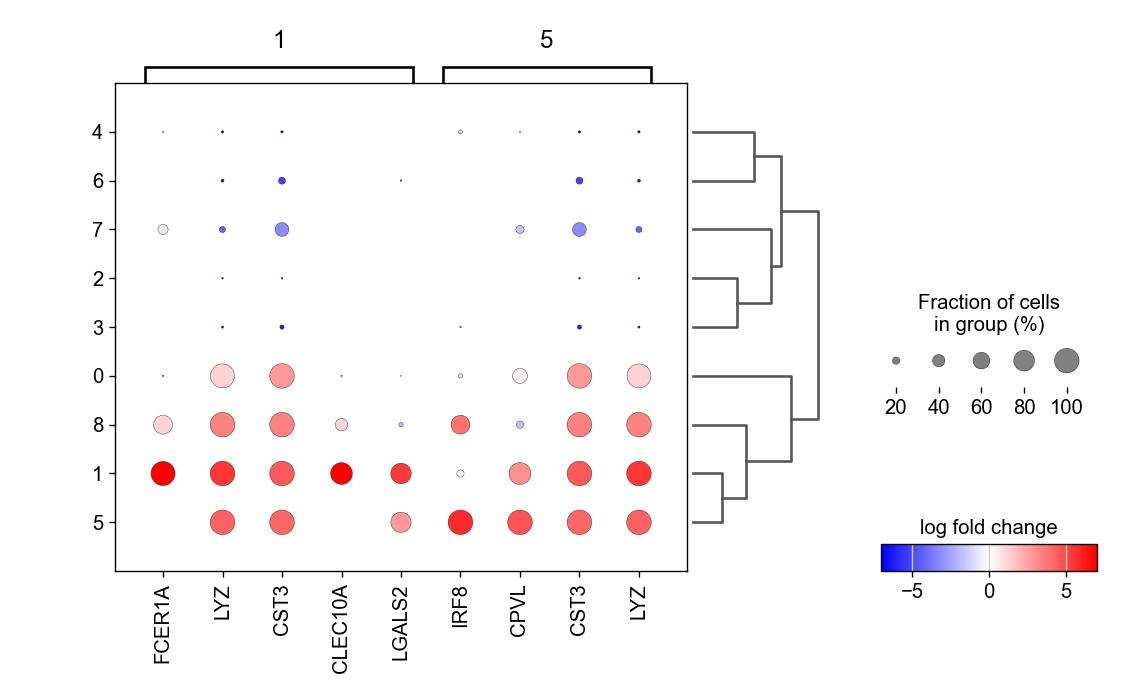
 下一步,我们集中关注不好分辨的两个组(簇1和5),在这里,我们增加参数`groups=['1', '5']`,并且设置更大范围的基因,再取对数。
下一步,我们集中关注不好分辨的两个组(簇1和5),在这里,我们增加参数`groups=['1', '5']`,并且设置更大范围的基因,再取对数。
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=30, values_to_plot='logfoldchanges',
min_logfoldchange=4, vmax=7, vmin=-7, cmap='bwr',
groups=['1', '5'])
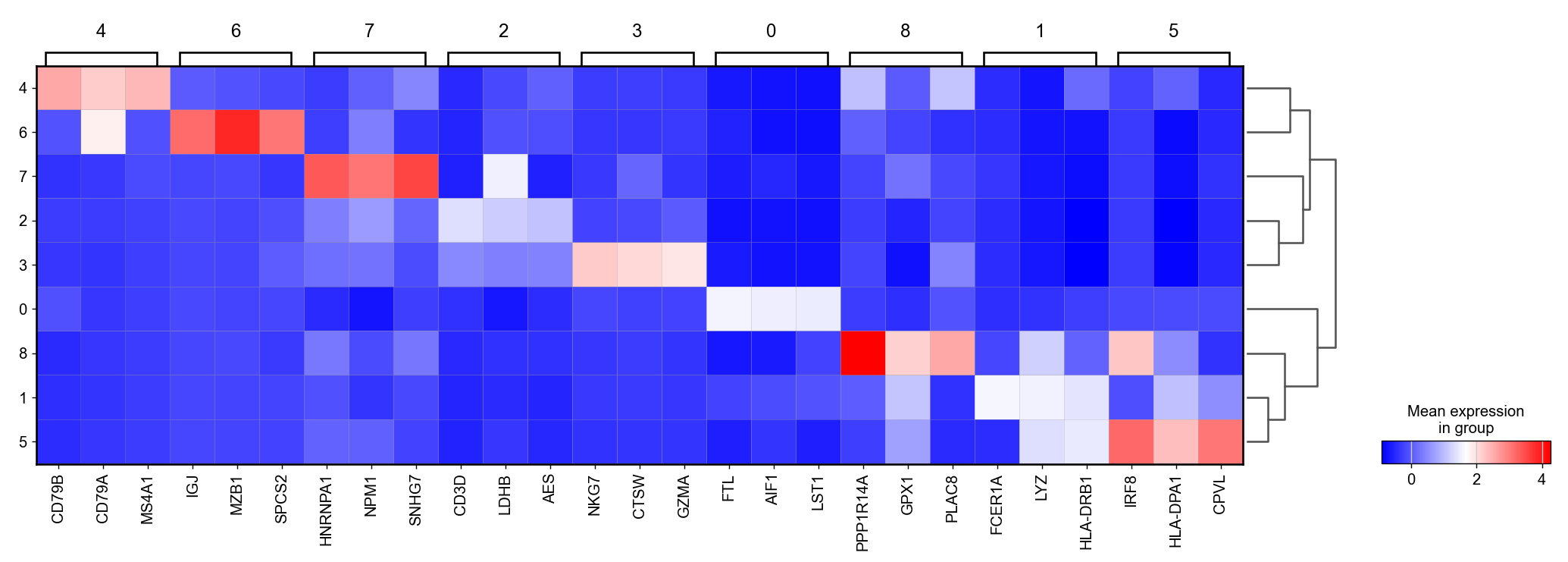
6.2 matrixplot可视化marker基因
对于下图,我们使用之前计算的scale数据(存储在layers下)可视化。
# `'bwr'` 是一种颜色映射方案,它从蓝色到白色再到红色的渐变,用于表示数值的正负变化。
sc.pl.rank_genes_groups_matrixplot(pbmc, n_genes=3, use_raw=False, cmap='bwr', layer='scaled')
参数解释:
n_genes=3:这个参数指定了要显示的排名前几个基因。在这里,n_genes=3表示显示排名前三个基因。use_raw=False:这个参数指定了是否使用原始数据来绘制矩阵图。在这里,use_raw=False表示不使用原始数据,而是使用经过处理的数据(例如标准化或归一化后的数据)来绘制矩阵图。cmap='bwr':这个参数指定了矩阵图的颜色映射。在这里,'bwr'是一种颜色映射方案,它从蓝色到白色再到红色的渐变,用于表示数值的正负变化。因此,较小的值会显示为蓝色,而较大的值会显示为红色。layer='scaled':这个参数指定了要绘制的数据层。在这里,'scaled'表示使用经过缩放处理的数据,即标准化或归一化后的数据。
6.3 tracksplot可视化marker基因
每个簇都选出差异得分前3的基因,下图右侧即为每个簇的前3高差异表达基因。
sc.pl.rank_genes_groups_tracksplot(pbmc, n_genes=3)
# 标准化或归一化后的数据
sc.pl.rank_genes_groups_tracksplot(pbmc, n_genes=3, use_raw=False, cmap='bwr', layer='scaled')

7. violin plot比较关键基因
在scanpy中,使用split violin plots同时对所有簇比较标记基因非常容易。我们对每个比较都展示前20个高差异得分的基因:
with rc_context({'figure.figsize': (9, 1.5)}):
sc.pl.rank_genes_groups_violin(pbmc, n_genes=20, jitter=False)
每个簇都与其他簇比较,会可视化得到9个子图(0-rest直到8-rest),比如0-rest:

我们可以对比上面两张图(tracksplot和split violin 0-rest),发现前3个差异基因正好是FTL,AIF1,LST1。
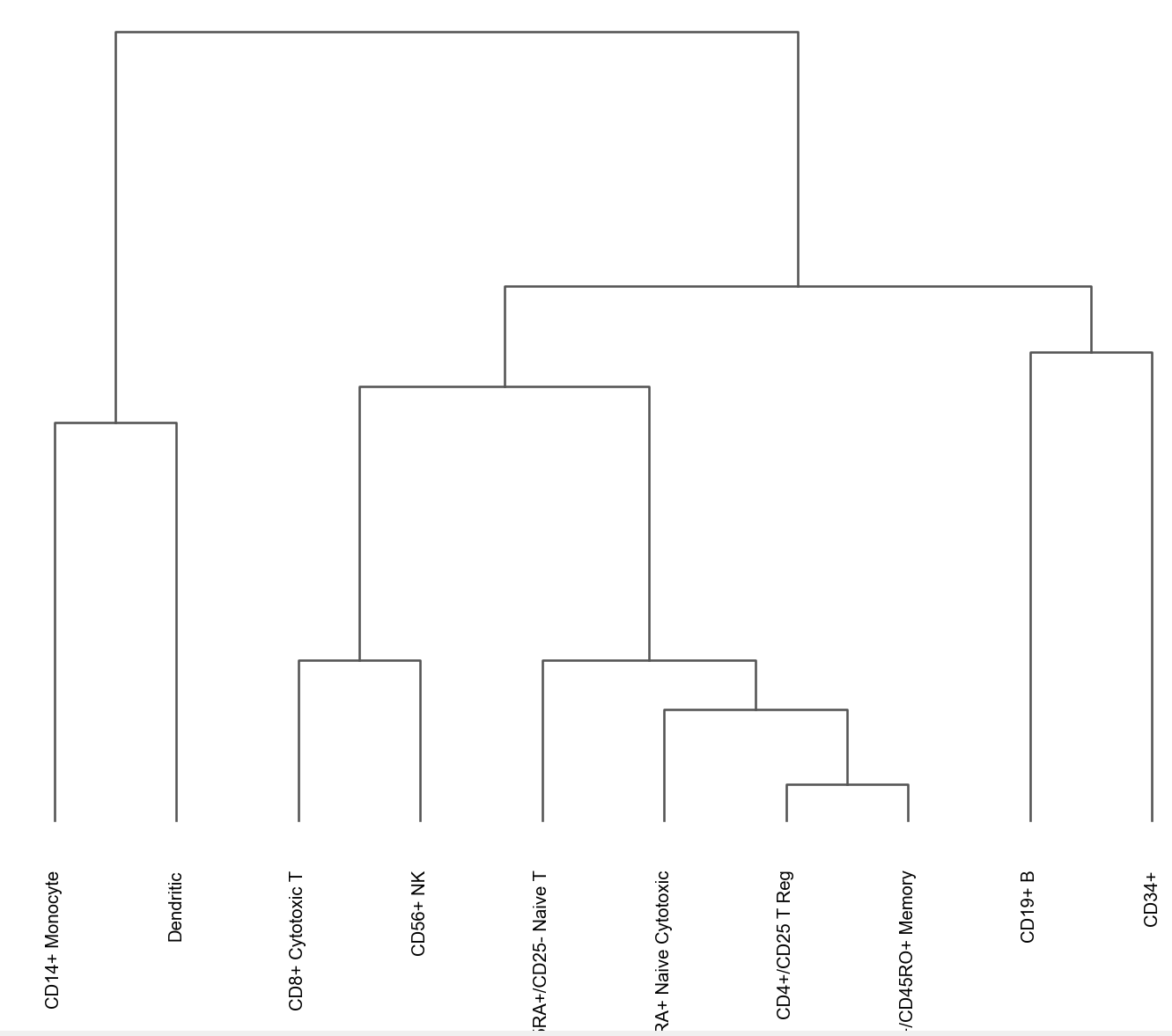
8. 树状图选项Dendrogram
进行样本的层次聚类分析,并将聚类结果可视化为聚类树状图
大多数可视化可以使用树状图来排列类别。当然,树状图也可以单独绘制,如下所示:
# 计算簇间的层次结构, 此处使用obs的bulk_labels做标签
sc.tl.dendrogram(pbmc, 'bulk_labels')
ax = sc.pl.dendrogram(pbmc, 'bulk_labels')
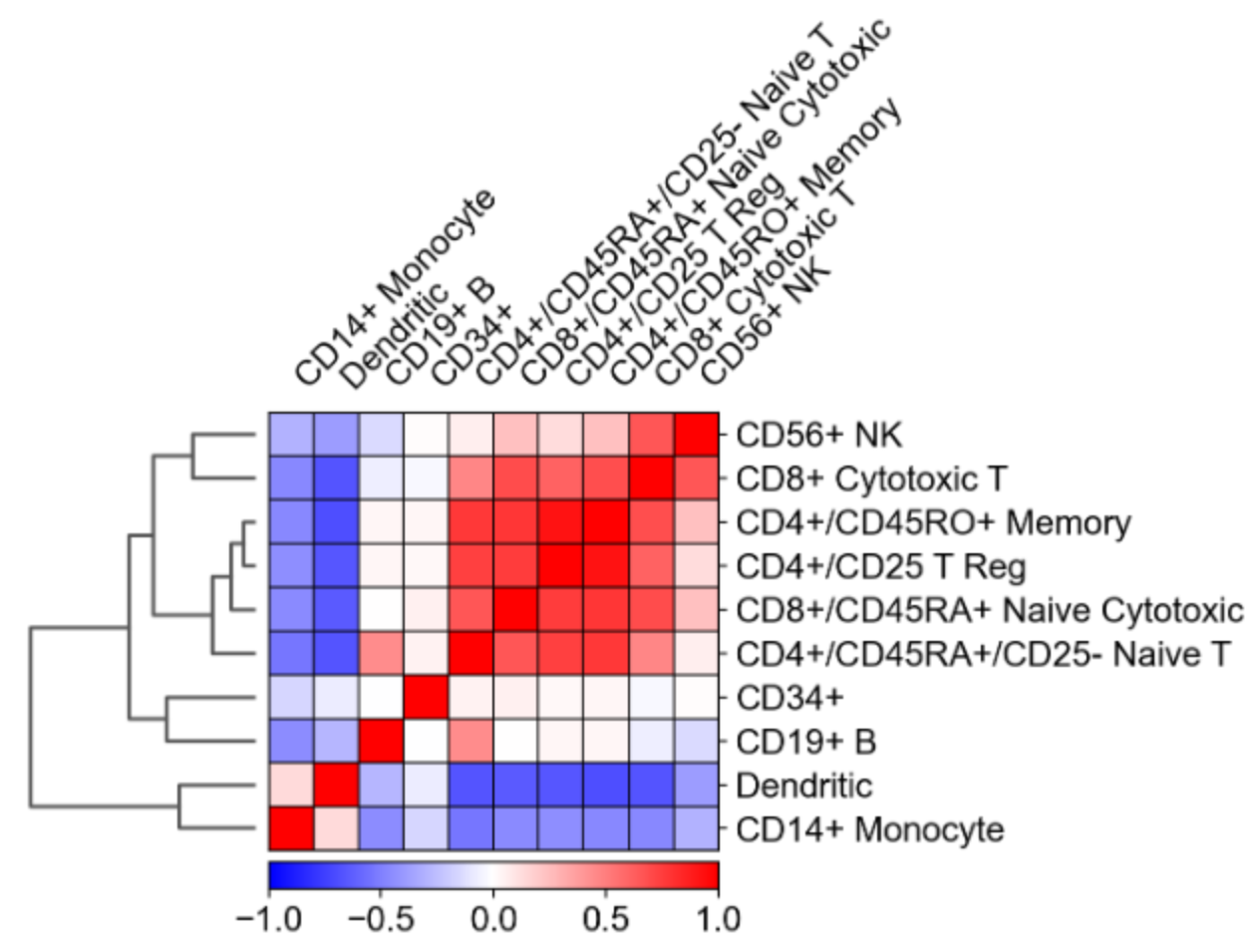
9. 相关性可视化
相关性的可视化可以与树状图一起,相关性可视化通常是绘制类别的相关性。
ax = sc.pl.correlation_matrix(pbmc, 'bulk_labels', figsize=(5,3.5))
1。
8. 树状图选项Dendrogram
进行样本的层次聚类分析,并将聚类结果可视化为聚类树状图
大多数可视化可以使用树状图来排列类别。当然,树状图也可以单独绘制,如下所示:
# 计算簇间的层次结构, 此处使用obs的bulk_labels做标签
sc.tl.dendrogram(pbmc, 'bulk_labels')
ax = sc.pl.dendrogram(pbmc, 'bulk_labels')
9. 相关性可视化
相关性的可视化可以与树状图一起,相关性可视化通常是绘制类别的相关性。
ax = sc.pl.correlation_matrix(pbmc, 'bulk_labels', figsize=(5,3.5))