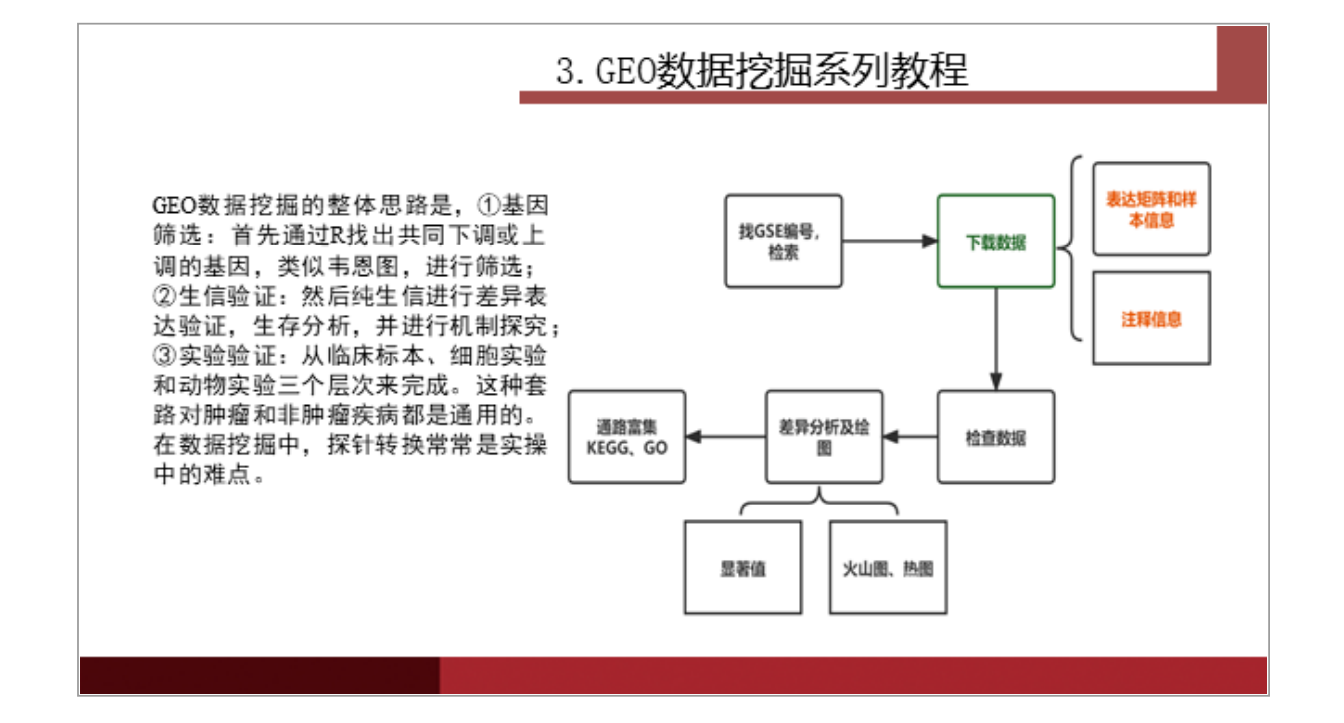
总言
两次作业汇报·其二:GEO数据处理学习汇报。
文章目录
- 总言
- 2、作业二:GEO数据处理下载分析
- 2.1、GEO数据库下载前准备
- 2.2、GEO数据库下载及数据初步处理
- 2.2.1、分阶段解析演示
- 2.2.1.1、编号下载流程
- 2.2.1.2、对gset[ 1 ]初步分析
- 2.2.1.3、对gset[ 2 ]初步分析
- 2.2.2、该部分整体脚本展示
- 2.3、以gset[1]中数据为例,进行数据检验
- 2.3.1、分阶段解析演示
- 2.3.1.1、绘制箱线图
- 2.3.1.2、PCA主成分分析
- 2.3.1.3、层次聚类分析
- 2.3.2、该部分整体脚本展示
- 2.4、以gset[1]中数据为例,将探针ID转换为基因ID
- 2.4.1、分阶段解析演示
- 2.4.2、该部分整体脚本展示
- 2.5、基于上述步骤,进行基因的差异性分析
- 2.5.1、分阶段解析演示
- 2.5.1.1、箱线图和t检验
- 2.5.1.2、差异分析走标准的limma流程
- 2.5.1.3、查看上调基因和下调基因
- 2.5.2、该部分整体脚本展示
- 2.6、基于上述步骤,绘制火山图、热图
- 2.6.1、分阶段解析演示
- 2.6.1.1、火山图
- 2.6.1.2、热图
- 2.6.2、该部分整体脚本展示
2、作业二:GEO数据处理下载分析
2.1、GEO数据库下载前准备
1)、NCBI:
相关链接
2)、GEO数据查看举例:
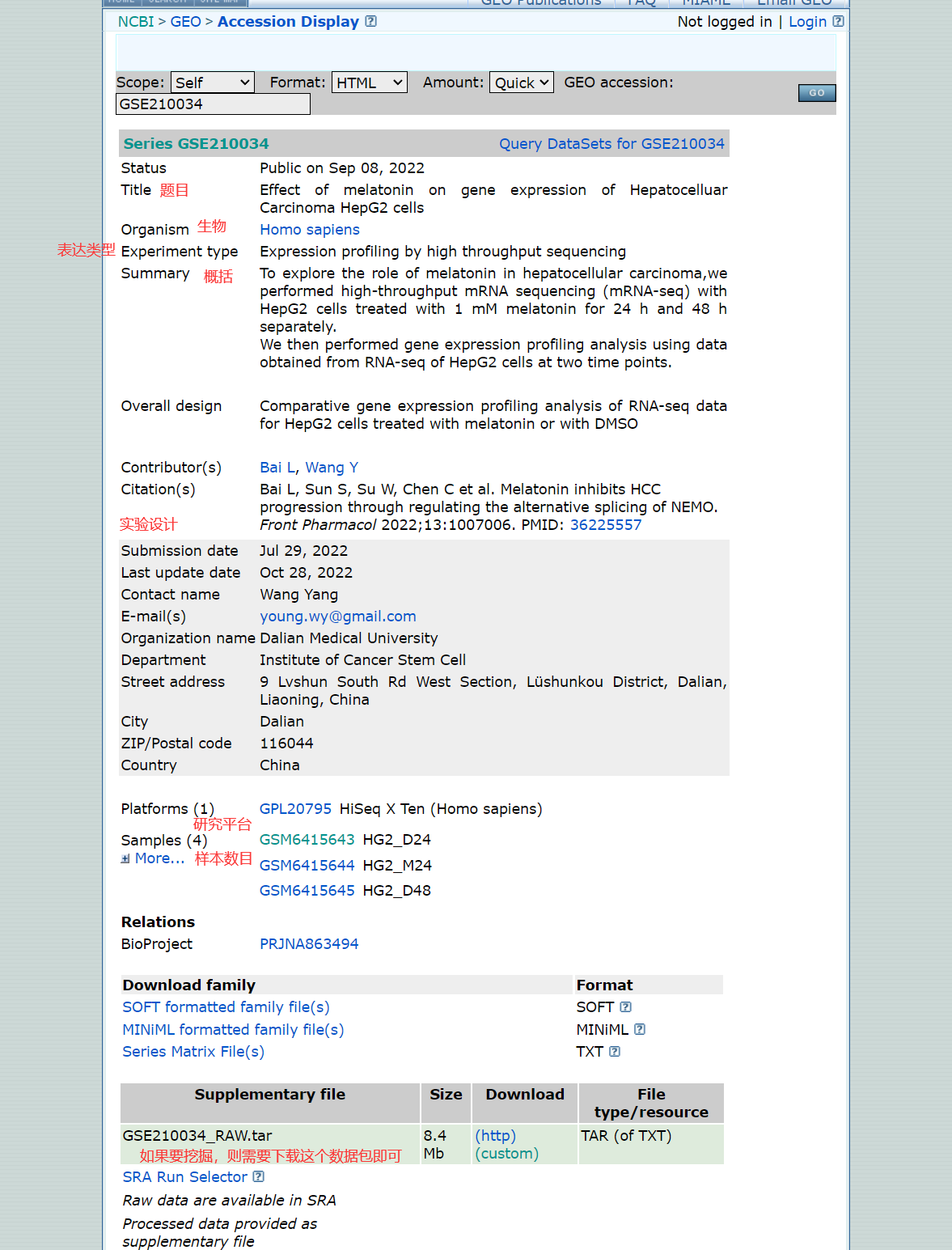
3)、演示数据说明:
①此处以参考资料中的GSE编码为例,其主要原因在于:比起随便寻找一条编码数据,该条编码比较方便演示后续数据分析的操作过程。
②编码号:GSE14520
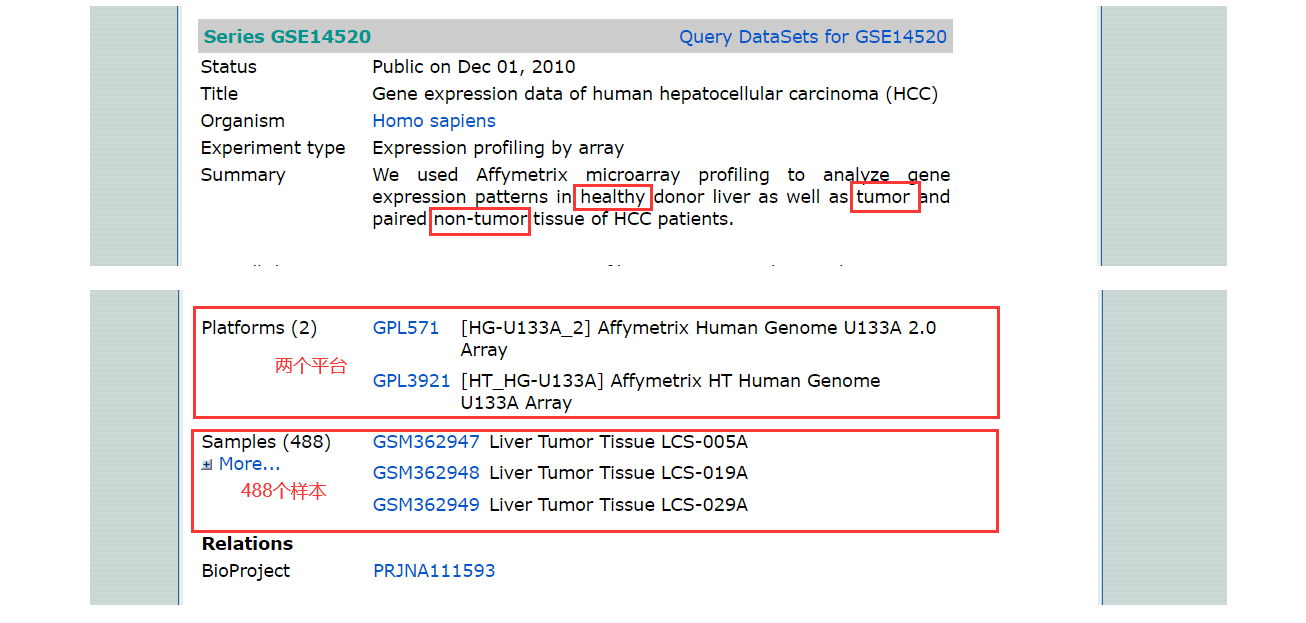
③该数据部分需要我们关注的信息展示:
2.2、GEO数据库下载及数据初步处理
对于相关数据下载,一种方式是在官网直接下载,此处我们采取的是:在RStudio使用相关包下载GEO数据。演示过程如下:
2.2.1、分阶段解析演示
2.2.1.1、编号下载流程
1)、选定编码号:
f='GSE14520_eSet.Rdata' ,同理更改GSE14520即可达到下载其它编码号的目的。
rm(list = ls()) ##该代码可用于清空environment中的相关数据。
options(stringsAsFactors = F) ##字符串是否作为因子?此处我们选择的是false。
f='GSE14520_eSet.Rdata' ##这里填写的是我们需要下载的GSE编号
2)、相关包的下载与加载:
要下载GEO数据库信息,需要下载GEOquery包,但其是包含在BiocManager包中的一个包,因此需要先下载BiocManager包。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")##下载"BiocManager"包
if (!requireNamespace("GEOquery", quietly = TRUE))
BiocManager::install("GEOquery")##下载"GEOquery"包
library(GEOquery) ##加载包
3)、下载GSE14520这个数据 :
利用GEOquery包中的getGEO函数来下载需要的GSE编号GSE14520。
#下载'GSE14520'这个数据
if(!file.exists(f)){
gset <- getGEO('GSE14520', destdir=".",
AnnotGPL = F, ##注释文件
getGPL = F) ##平台文件
save(gset,file=f) ##保存到本地
}
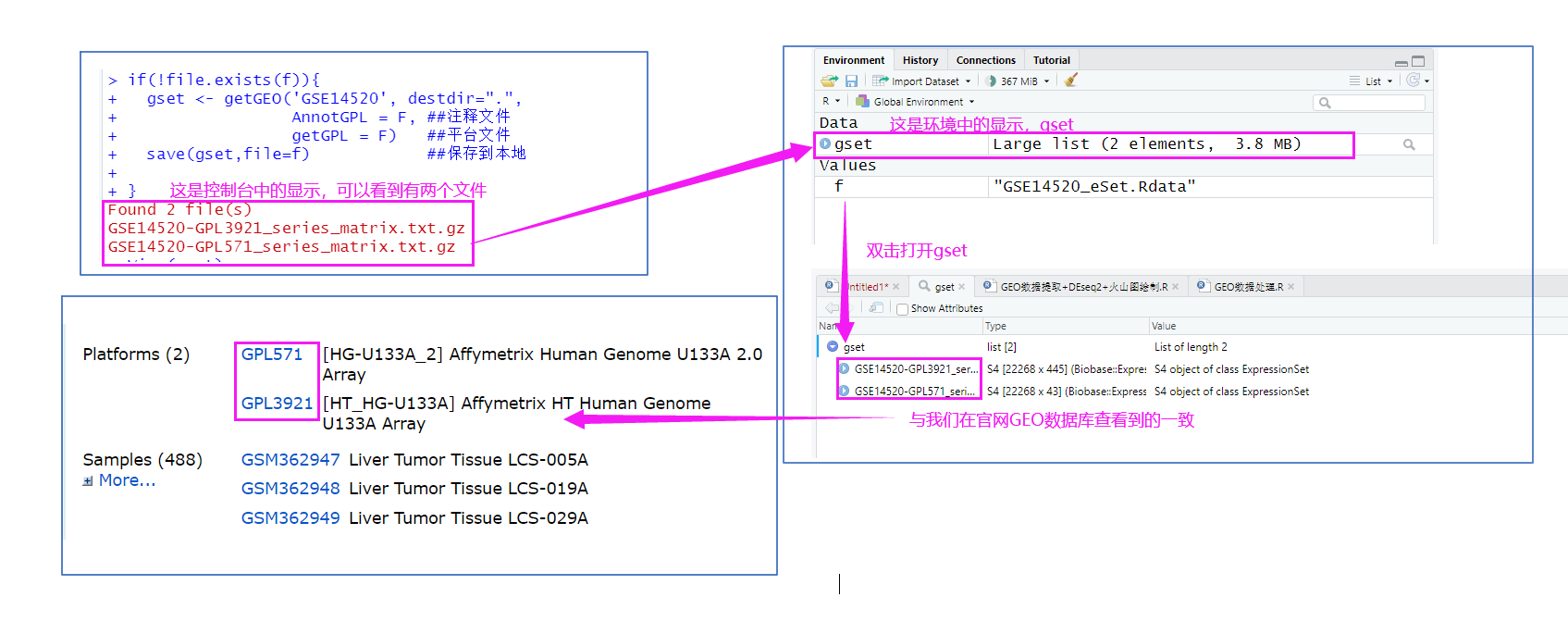
2.2.1.2、对gset[ 1 ]初步分析
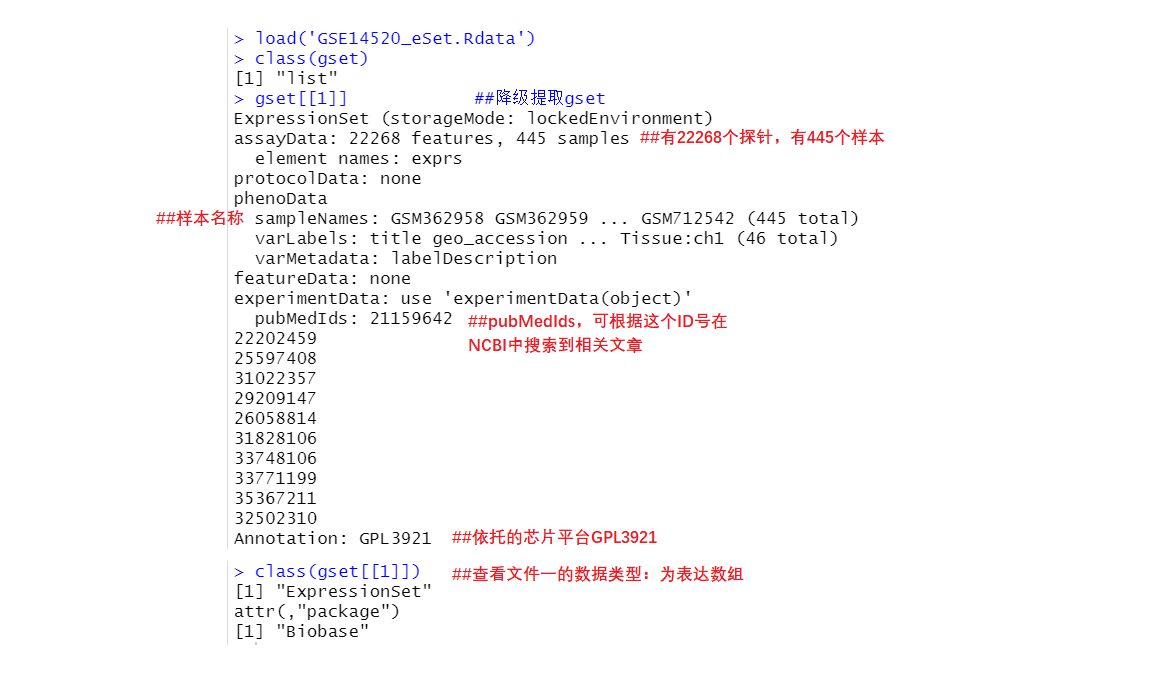
1)、降级提取gset[1] :
两个[[]]能达到提取文件的目的,gset[[1]] 表示提取第一个文件。
load('GSE14520_eSet.Rdata')
class(gset)
> class(gset) ##查看gset的类型
[1] "list"
> gset[[1]] ##降级提取gset
ExpressionSet (storageMode: lockedEnvironment) ##表达数组
assayData: 22268 features, 445 samples ##有22268个探针,有445个样本
element names: exprs
protocolData: none
phenoData
sampleNames: GSM362958 GSM362959 ... GSM712542 (445 total) ##样本名称
varLabels: title geo_accession ... Tissue:ch1 (46 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 21159642 ##pubMedIds,可根据这个ID号在NCBI中搜索到相关文章
22202459
25597408
31022357
29209147
26058814
31828106
33748106
33771199
35367211
32502310
Annotation: GPL3921 ##依托的芯片平台GPL3921
> class(gset[[1]]) ##查看文件一的数据类型:为表达数组
[1] "ExpressionSet"
attr(,"package")
[1] "Biobase"
2)、获取样本的表达矩阵,并将病人的临床信息按一定依据分组:
> dat1<-exprs(gset[[1]])##使用函数exprs获取样本表达矩阵
> pd1<-pData(gset[[1]]) ##使用函数pData获取样本临床信息(如性别、年龄、肿瘤分期等等)
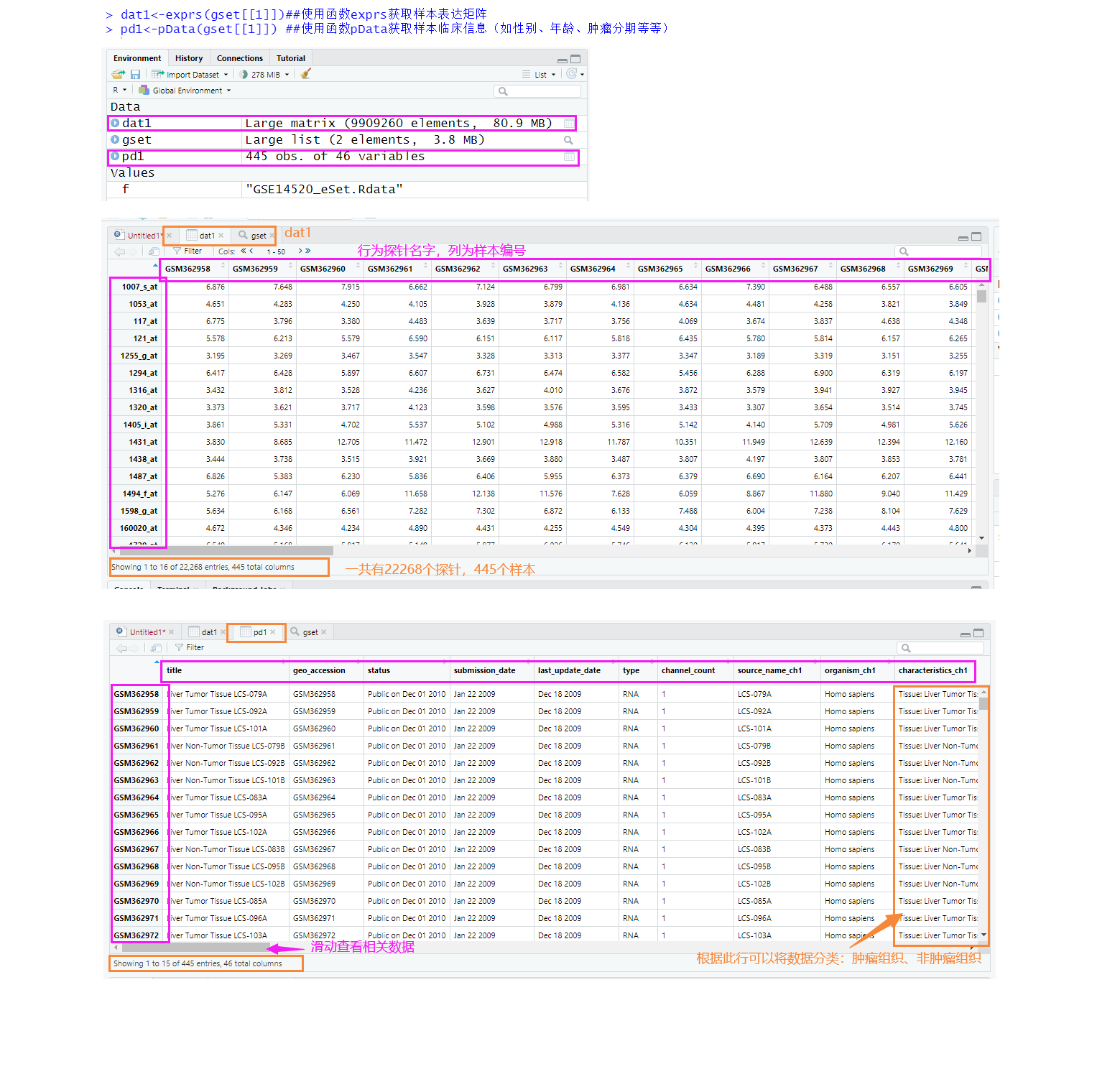
3)、分组,初步处理数据与保存:
根据2)中提供信息pd1$characteristics_ch1,我们根据是否为肿瘤组织('Tumor','Non_Tumor'),将数据样本分类存储于group_list1中,并将其保存save命名为Expreset1.Rdata,这样一来之后若还需要使用,就能直接在本地获取到相关数据。
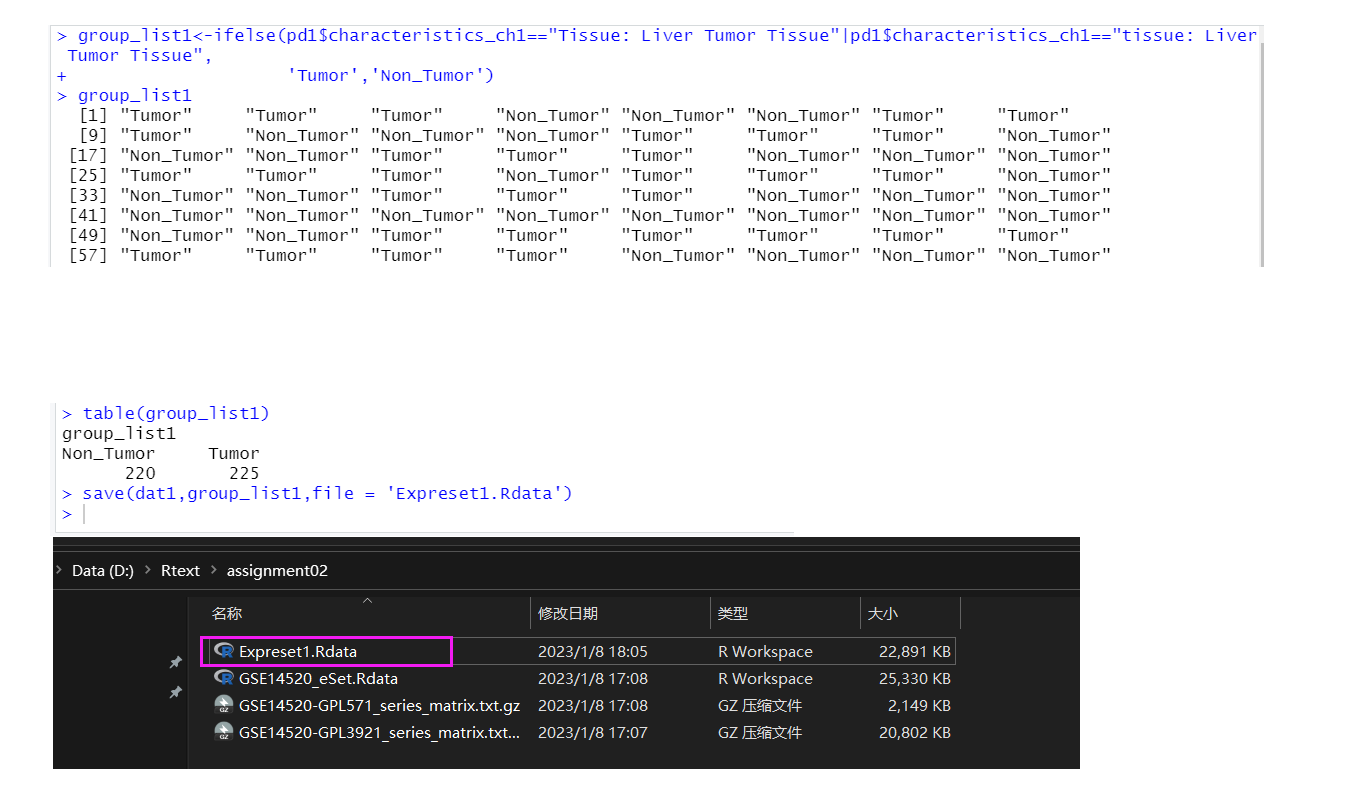
> group_list1<-ifelse(pd1$characteristics_ch1=="Tissue: Liver Tumor Tissue"|pd1$characteristics_ch1=="tissue: Liver Tumor Tissue",
+ 'Tumor','Non_Tumor')
> class(group_list1)
[1] "character"
> group_list1
[1] "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Tumor"
[9] "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor"
[17] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[25] "Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor"
[33] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[41] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[49] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[57] "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[65] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor"
[73] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[81] "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[89] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[97] "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[105] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor"
[113] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[121] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[129] "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[137] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[145] "Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[153] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor"
[161] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[169] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[177] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[185] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[193] "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[201] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[209] "Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[217] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[225] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[233] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[241] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Tumor"
[249] "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor"
[257] "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[265] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[273] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor"
[281] "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor"
[289] "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor"
[297] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[305] "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[313] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor"
[321] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[329] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[337] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[345] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor"
[353] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[361] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[369] "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor" "Tumor"
[377] "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor"
[385] "Non_Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[393] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor"
[401] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor"
[409] "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor"
[417] "Non_Tumor" "Non_Tumor" "Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor"
[425] "Tumor" "Tumor" "Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor"
[433] "Non_Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor"
[441] "Non_Tumor" "Non_Tumor" "Non_Tumor" "Non_Tumor" "Tumor"
如下,可看到Non_Tumor有220个,Tumor 有225个。
> table(group_list1)
group_list1
Non_Tumor Tumor
220 225
> save(dat1,group_list1,file = 'Expreset1.Rdata')
2.2.1.3、对gset[ 2 ]初步分析
同理,可对gset[[2]]初步处理,以下为演示过程:
1)、降级提取gset[2] :
> gset[[2]] ##同理,可对gset[[2]]初步处理,以下为演示过程
ExpressionSet (storageMode: lockedEnvironment)
assayData: 22268 features, 43 samples ##有22268个探针,43个样本
element names: exprs
protocolData: none
phenoData
sampleNames: GSM362947 GSM362948 ... GSM363451 (43 total)
varLabels: title geo_accession ... Tissue:ch1 (43 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 21159642 ##以下为pubMedIds
22202459
25597408
31022357
29209147
26058814
31828106
33748106
33771199
35367211
32502310
Annotation: GPL571 ##依托的平台
> class(gset[[2]])
[1] "ExpressionSet"
attr(,"package")
[1] "Biobase"
2)、获取样本的表达矩阵,并将病人的临床信息按一定依据分组:
> dat2<-exprs(gset[[2]])##使用函数exprs获取样本表达矩阵
> pd2<-pData(gset[[2]]) ##使用函数pData获取样本临床信息(如性别、年龄、肿瘤分期等等)
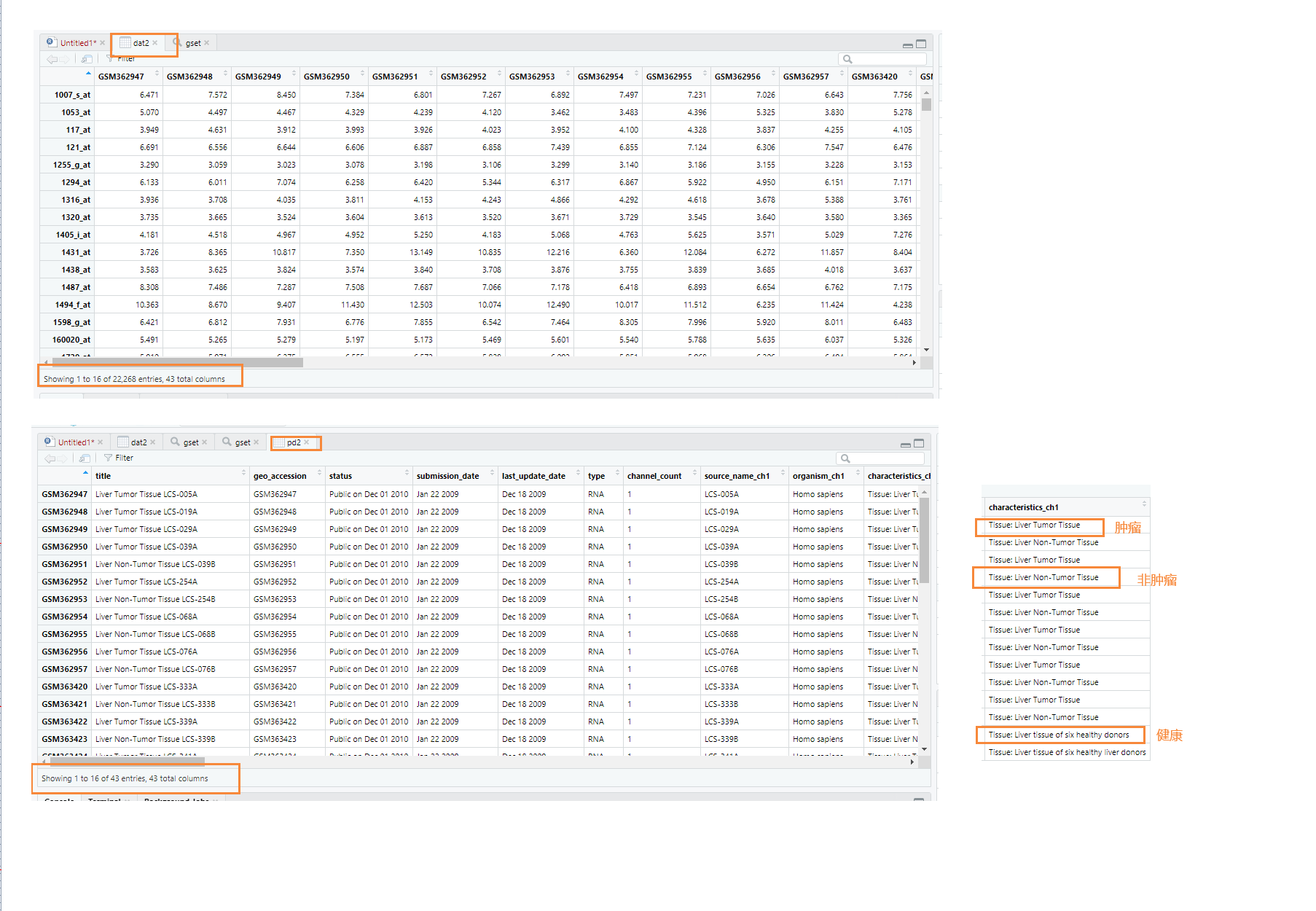
3)、分组,初步处理数据与保存:
如下,将病人分为了三类,Healthy有2个,Non_Tumor_HCC 有19个,Tumor_HCC 有22个。
> group_list2<-ifelse(pd2$characteristics_ch1=="Tissue: Liver Non-Tumor Tissue","Non_Tumor_HCC",
+ ifelse(pd2$characteristics_ch1=="Tissue: Liver Tumor Tissue","Tumor_HCC","Healthy"))
> table(group_list2)
group_list2
Healthy Non_Tumor_HCC Tumor_HCC
2 19 22
> save(dat2,group_list2,file = 'Expreset2.Rdata')
2.2.2、该部分整体脚本展示
###一、下载表达矩阵的信息以及初步数据处理
#####################################################
rm(list = ls()) ##该代码可用于清空environment中的相关数据。
options(stringsAsFactors = F) ##字符串是否作为因子?此处我们选择的是false。
f='GSE14520_eSet.Rdata' ##这里填写的是我们需要下载的GSE编号
##要下载GEO数据库信息,需要下载GEOquery包,但其是包含在BiocManager包中的一个包,因此需要先下载BiocManager包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")##下载"BiocManager"包
if (!requireNamespace("GEOquery", quietly = TRUE))
BiocManager::install("GEOquery")##下载"GEOquery"包
library(GEOquery)##加载包
##下载'GSE14520'这个数据
if(!file.exists(f)){
gset <- getGEO('GSE14520', destdir=".",
AnnotGPL = F, ##注释文件
getGPL = F) ##平台文件
save(gset,file=f) ##保存到本地
}
load('GSE14520_eSet.Rdata')
class(gset) ##查看gset的类型
gset[[1]] ##降级提取gset
class(gset[[1]]) ##查看文件一的数据类型:为表达数组
dat1<-exprs(gset[[1]])##使用函数exprs获取样本表达矩阵
pd1<-pData(gset[[1]]) ##使用函数pData获取样本临床信息(如性别、年龄、肿瘤分期等等)
group_list1<-ifelse(pd1$characteristics_ch1=="Tissue: Liver Tumor Tissue"|
pd1$characteristics_ch1=="tissue: Liver Tumor Tissue",'Tumor','Non_Tumor')
group_list1 ##查看group_list1
table(group_list1) ##整体汇总查看
save(dat1,group_list1,file = 'Expreset1.Rdata') ##将获取的这份信息输出保存
gset[[2]] ##同理,可对gset[[2]]初步处理,以下为演示过程
class(gset[[2]])
dat2<-exprs(gset[[2]])##使用函数exprs获取样本表达矩阵
pd2<-pData(gset[[2]]) ##使用函数pData获取样本临床信息(如性别、年龄、肿瘤分期等等)
group_list2<-ifelse(pd2$characteristics_ch1=="Tissue: Liver Non-Tumor Tissue","Non_Tumor_HCC",
ifelse(pd2$characteristics_ch1=="Tissue: Liver Tumor Tissue","Tumor_HCC","Healthy"))
table(group_list2)
save(dat2,group_list2,file = 'Expreset2.Rdata')
#####################################################
2.3、以gset[1]中数据为例,进行数据检验
1)、总述:
该步骤目的: 主要是为了检验我们下载的数据是否能够进行分析,以及初步展示数据情况。
主要方法:
①箱线图
②PCA主成分分析
③层次聚类分析
2.3.1、分阶段解析演示
2.3.1.1、绘制箱线图
1)、过程演示:
绘制箱线图时,考虑到Expreset1.Rdata是根据dat1生成的,而由上述内容其中含有442列数据,如果全部使用则绘制出来的箱线图特别密集,此处我们采取了前100数据作为观察dat1[,1:100]。
> rm(list = ls()) ##清空environment中的相关数据
> options(stringsAsFactors = F) ##取消字符串作为因子的设置
> load(file = 'Expreset1.Rdata') ##加载我们在步骤一中保存的Expreset1.Rdata数据
> boxplot(dat1[,1:100],cex=0.2,las=2,col="red") ##画箱线图
cex=0.2缩小到标准的0.2倍,col="red"颜色为红色。

①如图,由红色区域显示可知,用于观测的这100个样本的基因表达相对集中,在4~6之间,中位数也大致位于一条线上。
②若箱线图显示出的基因表达矩阵分布差距比较大,就需要对数据进行标准化、归一化。
2.3.1.2、PCA主成分分析
1)、简述:
PCA主成分分析主要目的是将数据进行降维处理。降维就是一种对高维度特征数据预处理方法。PCA主成分分析方法,是一种使用最广泛的数据降维算法。
以我们这次的数据为例,查看dat1时,我们统计了该数据探针数量为22268个,经过PCA降维处理,可将这些数据分为几类,然后我们分析这几类的表达差异即可。
2)、过程演示一:数据处理
①预处理:(非必要工作,视情况而定)
> rm(list = ls()) ##清空environment中的相关数据
> options(stringsAsFactors = F) ##取消字符串作为因子的设置
> load(file = 'Expreset1.Rdata') ##加载我们在步骤一中保存的Expreset1.Rdata数据
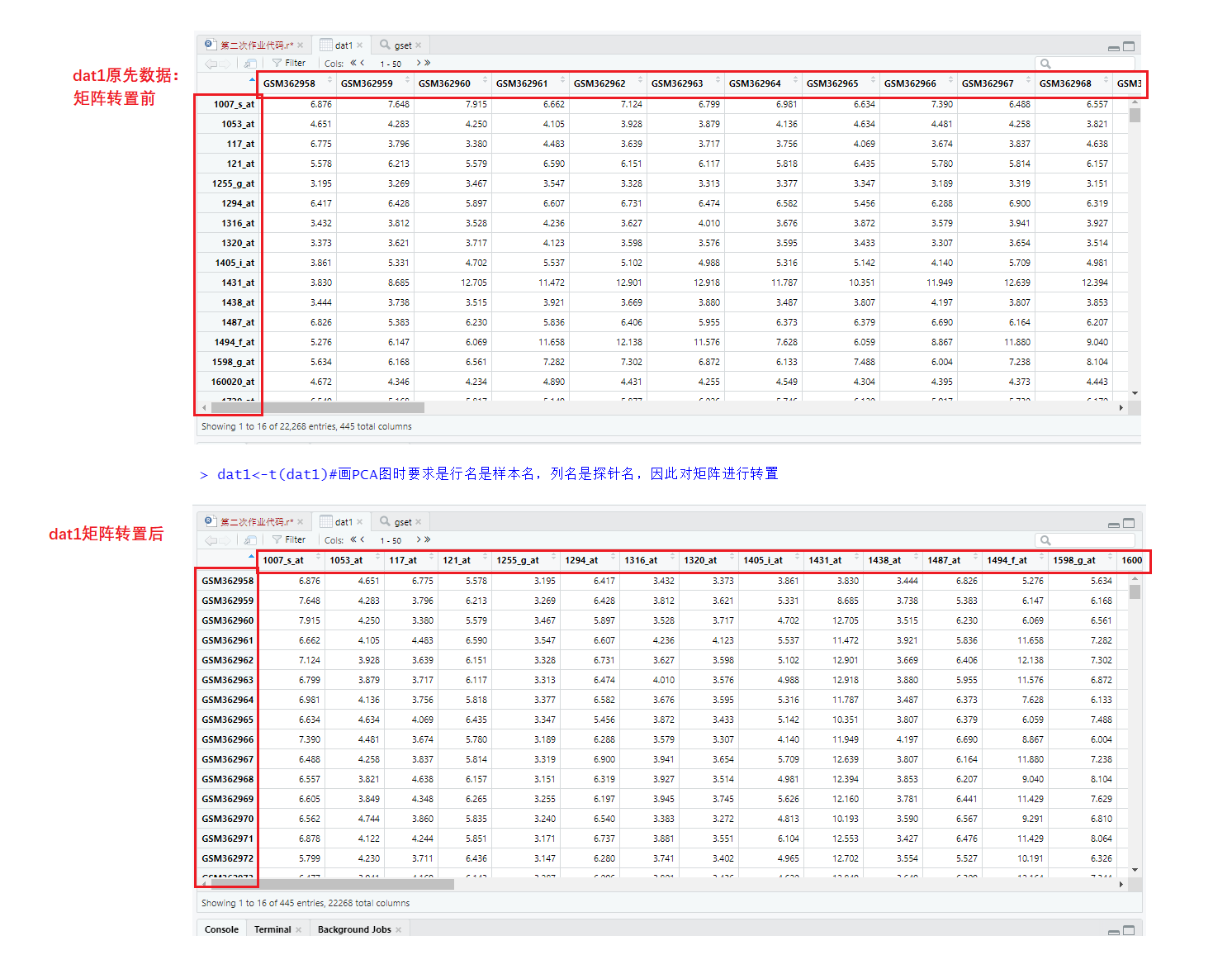
②在进行PCA主成分分析前,每次都要检测数据。如下,我们提取了矩阵的前四行、前四列检测。发现其行是GSM编号(样本名),列是探针名。
> dat1[1:4,1:4]##在进行PCA主成分分析前,每次都要检测数据
GSM362958 GSM362959 GSM362960 GSM362961
1007_s_at 6.876 7.648 7.915 6.662
1053_at 4.651 4.283 4.250 4.105
117_at 6.775 3.796 3.380 4.483
121_at 5.578 6.213 5.579 6.590
③画PCA图时,要求是行名是样本名,列名是探针名,因此我们需要对矩阵行转置。
> dat1<-t(dat1)##画PCA图时要求是行名是样本名,列名是探针名,因此对矩阵进行转置
> dat1[1:4,1:4]##查看是否转置成功
1007_s_at 1053_at 117_at 121_at
GSM362958 6.876 4.651 6.775 5.578
GSM362959 7.648 4.283 3.796 6.213
GSM362960 7.915 4.250 3.380 5.579
GSM362961 6.662 4.105 4.483 6.590
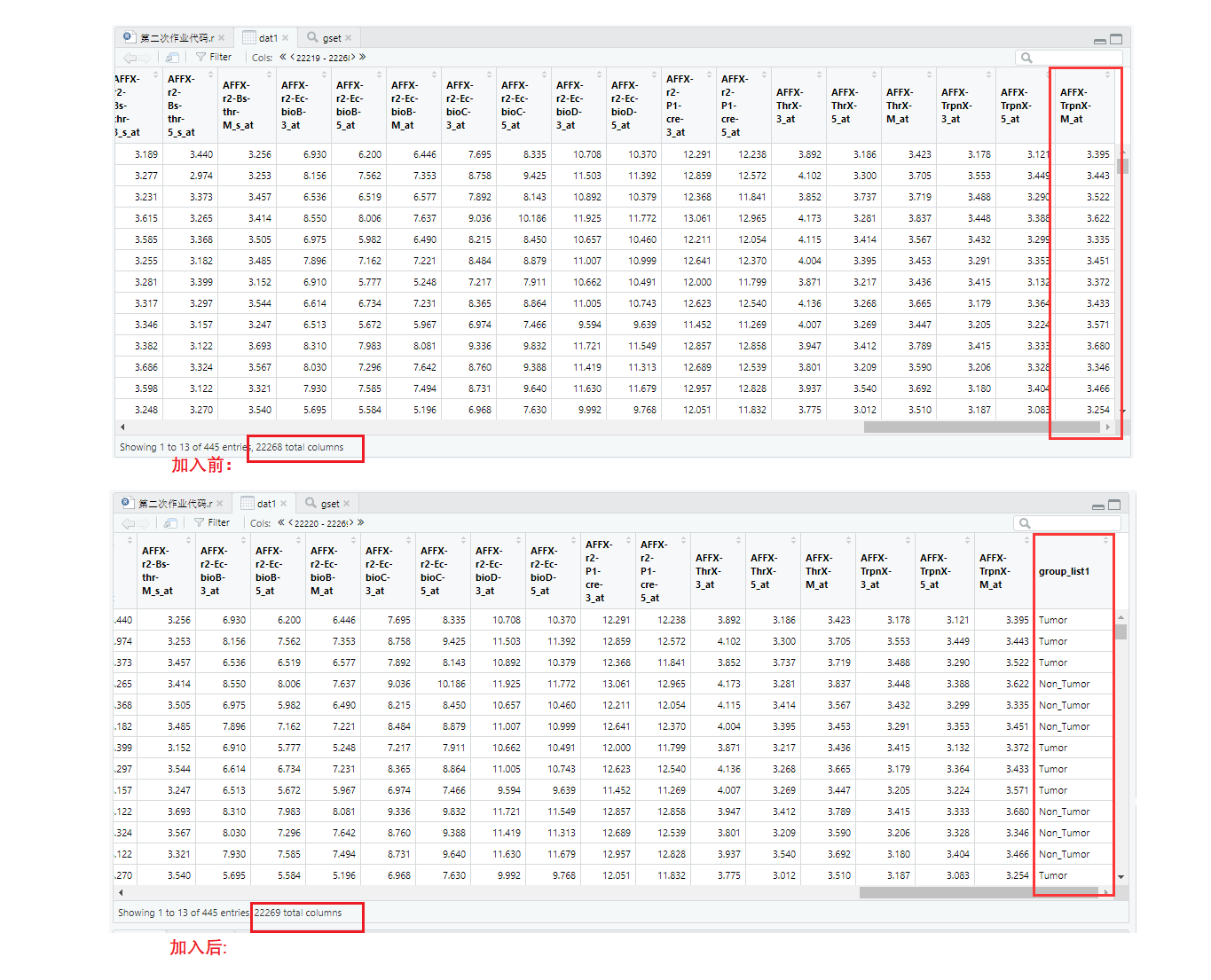
④将矩阵转换为数据框,并将分组信息(即'Tumor','Non_Tumor')作为最后一列加入dat1中。
> class(dat1);
[1] "matrix" "array"
> dat1<-as.data.frame(dat1)##将matrix转换为data.frame
> dat1<-cbind(dat1,group_list1)##cbind横向追加,即将分组信息group_list1追加到最后一列
> class(dat1);
[1] "data.frame"
3)、过程演示二:画图
①画主成分分析图需要加载"FactoMineR"、"factoextra"这两个包。

library("FactoMineR")##画主成分分析图需要加载这两个包
library("factoextra")
dat.pca <- PCA(dat1[,-ncol(dat1)], graph = FALSE)##将矩阵进行PCA分析,不包含最后一列(即我们的分组信息)
head(dat.pca$ind$coord)
用head查看以下我们进行的PCA分析结果,如下,它将数据分为了五个维度:
> head(dat.pca$ind$coord)
Dim.1 Dim.2 Dim.3 Dim.4 Dim.5
GSM362958 -37.72371 36.03526 3.130064 59.37616 -20.15968
GSM362959 -7.16702 62.49527 37.802374 34.30964 -37.75507
GSM362960 18.08779 68.40447 40.345610 57.98628 -23.63783
GSM362961 142.87247 45.84149 60.418633 50.58922 -55.68823
GSM362962 53.39154 -15.62183 14.978976 43.15879 -33.99242
GSM362963 74.05701 -11.75220 17.999868 42.29533 -25.63312
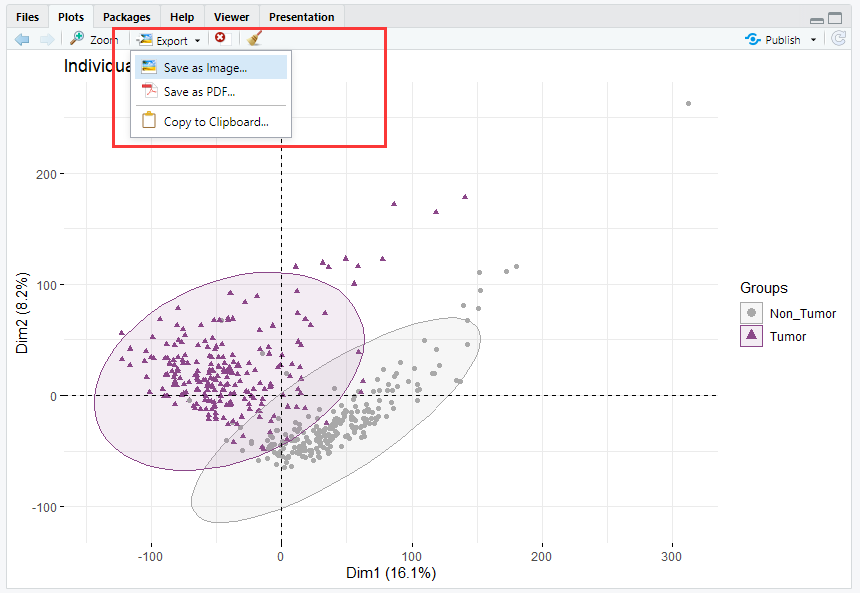
②以上述dat.pca结果画图:
> fviz_pca_ind(dat.pca,
+ geom.ind = "point", # show points only (nbut not "text")
+ col.ind = dat1$group_list1, # color by groups
+ palette = c("#00AFBB", "#E7B800"),
+ addEllipses = TRUE, # Concentration ellipses
+ legend.title = "Groups"
+ )

ggsave('Expreset1_PCA.png') ##这句代码可以将画出的图像以矢量形式存储。
也可以通过下述export直接导出。
2.3.1.3、层次聚类分析
聚类分析法(Cluster Analysis) 是在多元统计分析中研究如何对样品(或指标)进行分类的一种统计方法,它直接比较各事物之间的性质,将性质相近的归为一类,将性质差别较大的归入不同的类。
操作如下:
> rm(list = ls()) ##清空environment中的相关数据
> options(stringsAsFactors = F) ##取消字符串作为因子的设置
> load(file = 'Expreset1.Rdata') ##加载我们在步骤一中保存的Expreset1.Rdata数据
> dat1[1:4,1:4]##每次都要检测数据
GSM362958 GSM362959 GSM362960 GSM362961
1007_s_at 6.876 7.648 7.915 6.662
1053_at 4.651 4.283 4.250 4.105
117_at 6.775 3.796 3.380 4.483
121_at 5.578 6.213 5.579 6.590
层次聚类分析同样要求行名是样本名,列名是探针名,因此需要将矩阵转置
> dat1<-t(dat1)##层次聚类分析要求行名是样本名,列名是探针名,因此此时需要转置
> dat1[1:4,1:4]
1007_s_at 1053_at 117_at 121_at
GSM362958 6.876 4.651 6.775 5.578
GSM362959 7.648 4.283 3.796 6.213
GSM362960 7.915 4.250 3.380 5.579
GSM362961 6.662 4.105 4.483 6.590
以下是用d计算基因表达相近的样本的距离
> d<-dist(dat1)
> fit.complete<-hclust(d,method="complete")##hclust层次聚类
> plot(fit.complete,hang = -1,cex=0.8)##将聚类的结果绘制出来

2.3.2、该部分整体脚本展示
###二、数据的检验
#####################################################
##箱线图
rm(list = ls()) ##清空environment中的相关数据
options(stringsAsFactors = F)
load(file = 'Expreset1.Rdata')
boxplot(dat1[,1:100],cex=0.2,las=2,col="red")
##PCA主成分分析
rm(list = ls()) ##清空environment中的相关数据
options(stringsAsFactors = F)
load(file = 'Expreset1.Rdata')
dat1[1:4,1:4]##在进行PCA主成分分析前,每次都要检测数据
dat1<-t(dat1)##画PCA图时要求是行名是样本名,列名是探针名,因此对矩阵进行转置
dat1[1:4,1:4]##查看是否转置成功
class(dat1);
dat1<-as.data.frame(dat1)##将matrix转换为data.frame
dat1<-cbind(dat1,group_list1)##cbind横向追加,即将分组信息group_list1追加到最后一列
class(dat1);
library("FactoMineR")##画主成分分析图需要加载这两个包
library("factoextra")
dat.pca <- PCA(dat1[,-ncol(dat1)], graph = FALSE)##将矩阵进行PCA分析,不包含最后一列(即我们的)
head(dat.pca$ind$coord)
fviz_pca_ind(dat.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = dat1$group_list1, # color by groups
palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)
ggsave('Expreset1_PCA.png')
###层次聚类分析
rm(list = ls()) ##清空environment中的相关数据
options(stringsAsFactors = F) ##取消字符串作为因子的设置
load(file = 'Expreset1.Rdata') ##加载我们在步骤一中保存的Expreset1.Rdata数据
dat1[1:4,1:4]##每次都要检测数据
dat1<-t(dat1)##层次聚类分析要求行名是样本名,列名是探针名,因此需要将矩阵转置
dat1[1:4,1:4]
##此处是用d计算基因表达相近的样本的距离
d<-dist(dat1)
fit.complete<-hclust(d,method="complete")##hclust层次聚类
plot(fit.complete,hang = -1,cex=0.8)##将聚类的结果绘制出来
##扩充:
##使用factoextra,igraph包对它进行美化,首先画一张基础的分类图
library(factoextra)
library(igraph)
fviz_dend(fit.complete)##基础图
heatmap(as.matrix(d))##热图
#####################################################
2.4、以gset[1]中数据为例,将探针ID转换为基因ID
在上述过程中,我们知道dat1数据行名为探针名称,而我们并不知道它所对应的基因名称。故我们需要将探针的ID号转换为基因ID号。
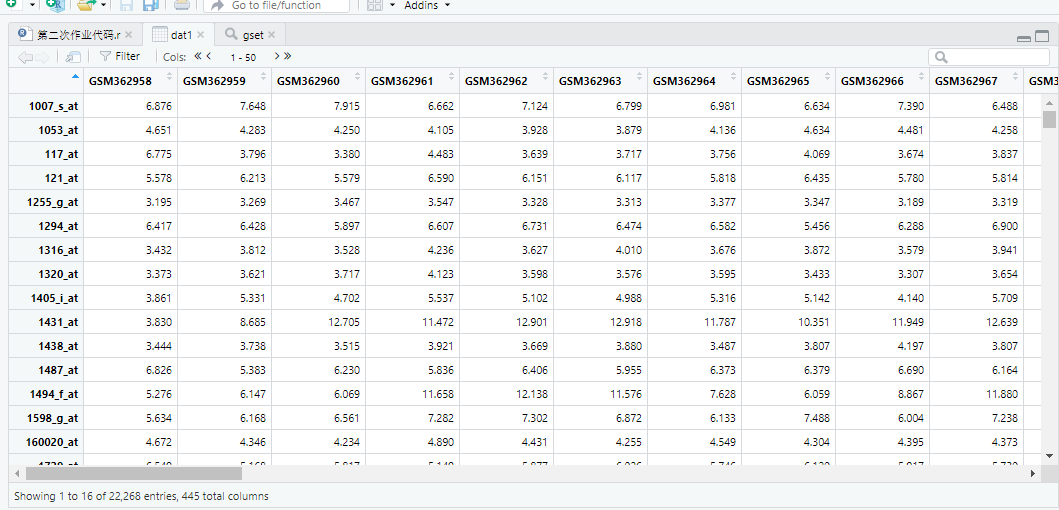
2.4.1、分阶段解析演示
①预处理:
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'Expreset1.Rdata')
②要想将探针的ID号转换为基因的ID号,需要知道所依托的芯片平台。
> gset[[1]] ##一中我们可用于降维提取时使用过,此处用来查看芯片平台
ExpressionSet (storageMode: lockedEnvironment)
assayData: 22268 features, 445 samples
element names: exprs
protocolData: none
phenoData
sampleNames: GSM362958 GSM362959 ... GSM712542 (445 total)
varLabels: title geo_accession ... Tissue:ch1 (46 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 21159642
22202459
25597408
31022357
29209147
26058814
31828106
33748106
33771199
35367211
32502310
Annotation: GPL3921
③如上图所示,其GPL平台是GPL3921,我们需要提取GPL3921芯片平台的探针ID和对应的基因,这里我们借助了相关的R包。而对这些R包的整理,此处附上一个网址:用R获取芯片探针与基因的对应关系三部曲
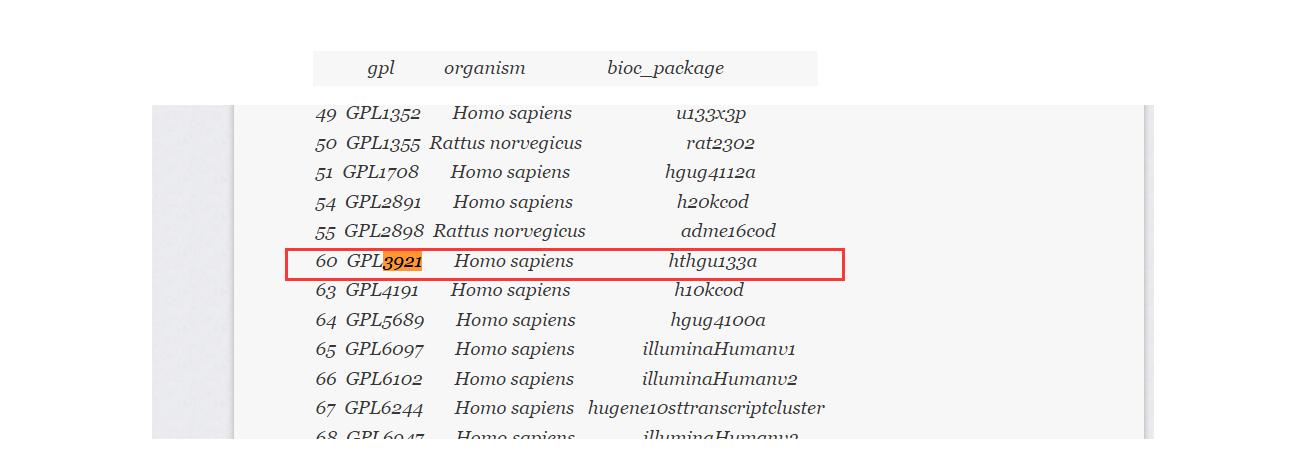
查询可得需要下载"hthgu133a.db"包,以下为相关代码,若"BiocManager"包没下载也需要一并下载,因为前者依托于后者中。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")##下载"BiocManager"包,注意.db
if (!requireNamespace("hthgu133a.db", quietly = TRUE))
BiocManager::install("hthgu133a.db")##下载"hthgu133a.db"包
下载好后需要提取GPL3921芯片平台的探针ID和对应的基因。
> library(hthgu133a.db)
> ls("package:hthgu133a.db")##查看包中相关数据
[1] "hthgu133a"
[2] "hthgu133a.db"
[3] "hthgu133a_dbconn"
[4] "hthgu133a_dbfile"
[5] "hthgu133a_dbInfo"
[6] "hthgu133a_dbschema"
[7] "hthgu133aACCNUM"
[8] "hthgu133aALIAS2PROBE"
[9] "hthgu133aCHR"
[10] "hthgu133aCHRLENGTHS"
[11] "hthgu133aCHRLOC"
[12] "hthgu133aCHRLOCEND"
[13] "hthgu133aENSEMBL"
[14] "hthgu133aENSEMBL2PROBE"
[15] "hthgu133aENTREZID"
[16] "hthgu133aENZYME"
[17] "hthgu133aENZYME2PROBE"
[18] "hthgu133aGENENAME"
[19] "hthgu133aGO"
[20] "hthgu133aGO2ALLPROBES"
[21] "hthgu133aGO2PROBE"
[22] "hthgu133aMAP"
[23] "hthgu133aMAPCOUNTS"
[24] "hthgu133aOMIM"
[25] "hthgu133aORGANISM"
[26] "hthgu133aORGPKG"
[27] "hthgu133aPATH"
[28] "hthgu133aPATH2PROBE"
[29] "hthgu133aPFAM"
[30] "hthgu133aPMID"
[31] "hthgu133aPMID2PROBE"
[32] "hthgu133aPROSITE"
[33] "hthgu133aREFSEQ"
[34] "hthgu133aSYMBOL"
[35] "hthgu133aUNIPROT"
ids <- toTable(hthgu133aSYMBOL)##提取GPL3921芯片平台的探针ID和对应的基因:SYMBOL是我们需要的注释信息ids
④现在就可以将dat1中探针ID变为基因名,演示如下:
> dat1<-as.data.frame(dat1)##先把表达矩阵变为数据框
> dat1$probe_id<-rownames(dat1)##将探针ID添加到dat1表达矩阵的新的一列
> dat1<-merge(dat1,ids,by='probe_id')#通过merge()函数,将dat1的探针id与芯片平台探针id相匹配,合并到dat1
⑤我们查看以下合成的dat1,最后一列为symbol,说明我们成功合并基因名称。但能发现的是,其中一些探针的基因名称是重复的。因此需要对多个探针的基因做处理。
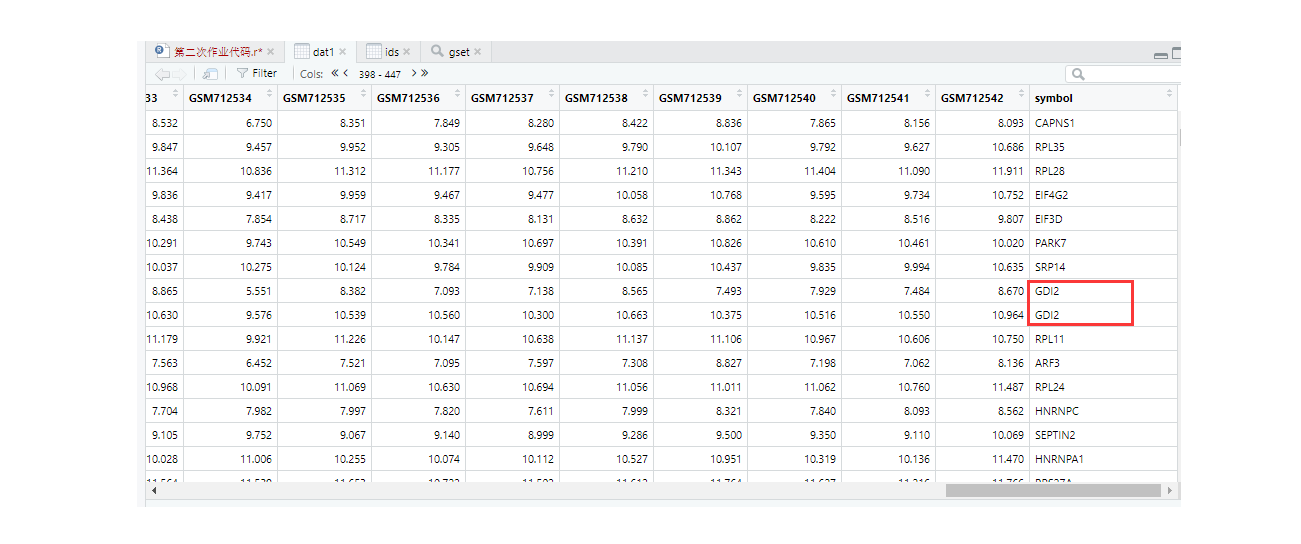
> library(limma)##取平均值需要limma这个包
> dat1<-avereps(dat1[,-c(1,447)],ID=dat1$symbol)
> dat1<-as.matrix(dat1)##转换
> save(dat1,group_list1,file = 'Expreset3.Rdata')
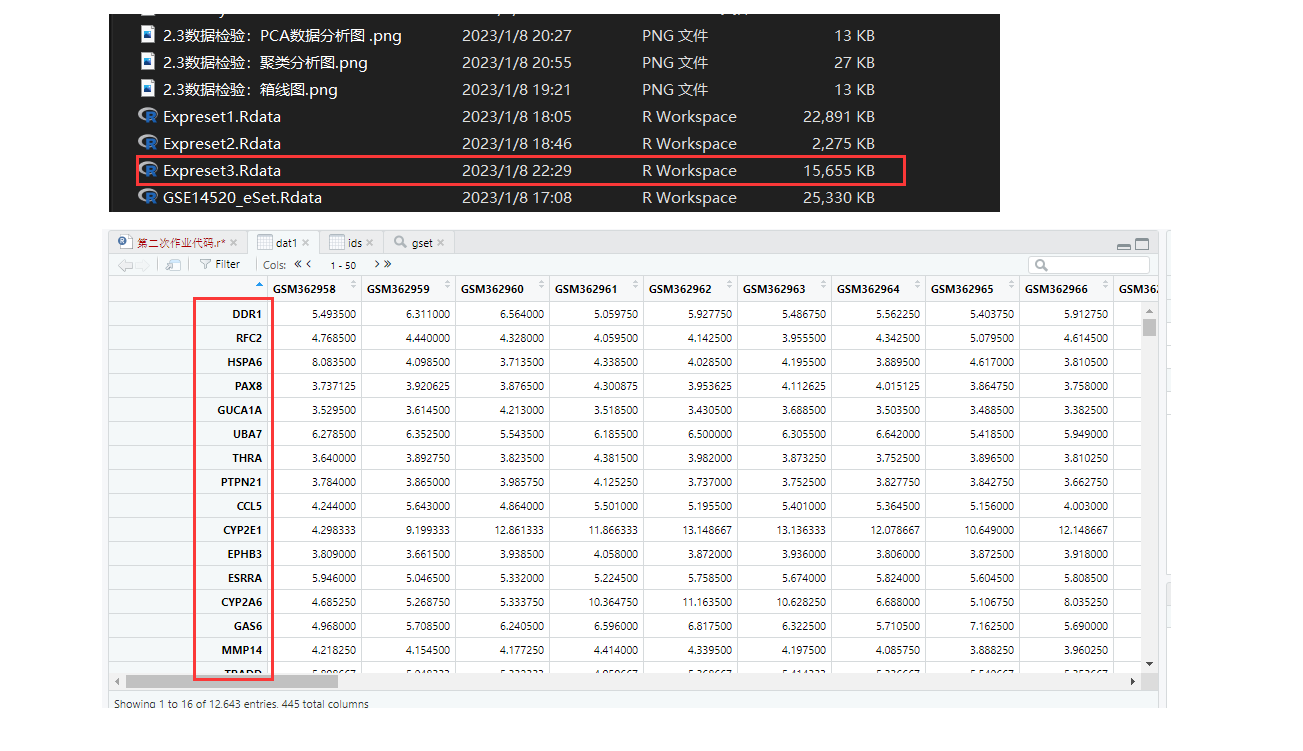
2.4.2、该部分整体脚本展示
###三、探针ID转换为基因ID
#####################################################
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'Expreset1.Rdata')
gset[[1]] ##一中我们可用于降维提取时使用过,此处用来查看芯片平台
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")##下载"BiocManager"包
if (!requireNamespace("hthgu133a.db", quietly = TRUE))
BiocManager::install("hthgu133a.db")##下载"hthgu133a.db"包
library(hthgu133a.db)
ls("package:hthgu133a.db")##查看相关数据
ids <- toTable(hthgu133aSYMBOL)##提取GPL3921芯片平台的探针ID和对应的基因:SYMBOL是我们需要的注释信息ids
dat1<-as.data.frame(dat1)##先把表达矩阵变为数据框
dat1$probe_id<-rownames(dat1)##将探针ID添加到dat1表达矩阵的新的一列
dat1<-merge(dat1,ids,by='probe_id')#通过merge()函数,将dat1的探针id与芯片平台探针id相匹配,合并到dat1
#多个探针检测一个基因,合并一起,取其平均值
library(limma)##取平均值需要limma这个包
dat1<-avereps(dat1[,-c(1,447)],ID=dat1$symbol)
dat1<-as.matrix(dat1)##转换
save(dat1,group_list1,file = 'Expreset3.Rdata')
#####################################################
2.5、基于上述步骤,进行基因的差异性分析
2.5.1、分阶段解析演示
2.5.1.1、箱线图和t检验
①要注意加载正确的表达矩阵,此处需要的是2.5章节中转换后的dat1。
> rm(list = ls()) ##一键清空
> options(stringsAsFactors = F)
> load(file = 'Expreset3.Rdata')##注意此处需要用探针ID转换为基因ID后的数据
> dat1[1:4,1:4]##每次都要检测数据:可以看到行名为基因名
GSM362958 GSM362959 GSM362960 GSM362961
DDR1 5.493500 6.311000 6.5640 5.059750
RFC2 4.768500 4.440000 4.3280 4.059500
HSPA6 8.083500 4.098500 3.7135 4.338500
PAX8 3.737125 3.920625 3.8765 4.300875
> table(group_list1) #table函数,查看group_list1中的分组个数
group_list1
Non_Tumor Tumor
220 225
②对基因以group_list分组画箱线图
Ⅰ、第一个基因:由下述代码可知,第一个基因Non_Tumor平均值为4.336030 ,Tumor 平均值为4.974918 。
> boxplot(dat1[1,]~group_list1) ##一个基因以group_list分组画箱线图
> t.test(dat1[1,]~group_list1) ##对第一个基因做t检验
Welch Two Sample t-test
data: dat1[1, ] by group_list1
t = -5.0617, df = 337.1, p-value = 6.849e-07
alternative hypothesis: true difference in means between group Non_Tumor and group Tumor is not equal to 0
95 percent confidence interval:
-0.5000559 -0.2201685
sample estimates:
mean in group Non_Tumor mean in group Tumor
5.352051 5.712163

Ⅱ、第二个基因:由下述代码可知,第二个基因Non_Tumor平均值为4.336030 ,Tumor 平均值为4.974918 。
> boxplot(dat1[2,]~group_list1) ##第二个基因以group_list分组画箱线图
> t.test(dat1[2,]~group_list1) ##对第二个基因做t检验
Welch Two Sample t-test
data: dat1[2, ] by group_list1
t = -15.524, df = 299.17, p-value < 2.2e-16
alternative hypothesis: true difference in means between group Non_Tumor and group Tumor is not equal to 0
95 percent confidence interval:
-0.7198772 -0.5578993
sample estimates:
mean in group Non_Tumor mean in group Tumor
4.336030 4.974918

Ⅲ、自定义一个函数,用于上述重复性操作,其中做了数据美化处理。
> bp=function(g){ ##自定义一个函数bp,函数为{}里的内容
+ library(ggpubr)
+ df=data.frame(gene=g,stage=group_list1)
+ p <- ggboxplot(df, x = "stage", y = "gene",
+ color = "stage", palette = "jco",
+ add = "jitter")
+ # Add p-value
+ p + stat_compare_means()
+ }
> bp(dat1[3,]) ## 调用上面定义好的函数,避免同样的绘图代码重复多次敲。
##同理可尝试其它:
##bp(dat1[1,])
##bp(dat1[2,])
##bp(dat1[4,])
##……

2.5.1.2、差异分析走标准的limma流程
1)、概述:
由于上述2.5.1.1中的差异化分析是一个基因一个基因进行的,相对比较低效麻烦,此处我们可以直接对整体的数据进行差异化分析,则需要走limma流程。
走limma流程前首先要准备的相关数据:
①表达矩阵:上述步骤中的dat1即是。
②分组矩阵:我们没有,需要创建。
③差异比较矩阵:我们没有,也需要创建。
2)、分组矩阵创建:
代码如下:
##差异分析走标准的limma流程
> library(limma)
> design=model.matrix(~0+factor(group_list1))##创建一个分组的矩阵
> colnames(design)=levels(factor(group_list1))
> rownames(design)=colnames(dat1)
> head(design)
Non_Tumor Tumor
GSM362958 0 1
GSM362959 0 1
GSM362960 0 1
GSM362961 1 0
GSM362962 1 0
GSM362963 1 0
分组矩阵中,将矩阵分为Non_Tumor、Tumor两个部分,0表示否/false,1表示是/true。
例如上述GSM362958 ,Non_Tumor=0即其不是非肿瘤组,Tumor=1,即其是肿瘤组。
3)、差异比较矩阵创建:
代码如下:
> ##创建差异比较矩阵
> contrast.matrix<-makeContrasts(paste0(unique(group_list1),collapse = "-"),levels = design)
> contrast.matrix ##这个矩阵声明,我们要Tumor组和Non_Tumor组进行差异分析比较
Contrasts
Levels Tumor-Non_Tumor
Non_Tumor -1
Tumor 1
4)、limma流程三板斧:
①第一步lmFit:lmFit为每个基因给定一系列的阵列来拟合线性模型
> ##limma流程三板斧:
> ##第一步lmFit
> ##lmFit为每个基因给定一系列的阵列来拟合线性模型
> fit<-lmFit(dat1,design)
②第二步eBayes:eBayes给出了一个微阵列线性模型拟合,通过经验贝叶斯调整标准误差到一个共同的值来计算修正后的t统计量、修正后的f统计量和微分表达式的对数概率。
> ##第二步eBayes
> ##eBayes给出了一个微阵列线性模型拟合,
> ##通过经验贝叶斯调整标准误差到一个共同的值来计算修正后的t统计量、修正后的f统计量和微分表达式的对数概率。
> fit2<-contrasts.fit(fit, contrast.matrix)
> fit2<-eBayes(fit2)
③第三步topTable:topTable是从线性模型拟合中提取出排名靠前的基因表。
> ##第三步topTable
> ##topTable是从线性模型拟合中提取出排名靠前的基因表。
> options(digits = 4) #设置全局的数字有效位数为4
> ##topTable(fit2,coef=2,adjust='BH')
然后我们就得到的相关数据:
> tempOutput<-topTable(fit2,coef=1,n=Inf)
> tempOutput<-na.omit(tempOutput)##如果有NA值,则需要移除
> head(tempOutput)
logFC AveExpr t P.Value adj.P.Val B
ECM1 -2.378 6.391 -43.73 2.164e-163 2.736e-159 363.0
VIPR1 -2.853 5.699 -41.68 6.499e-156 4.108e-152 345.9
CXCL14 -3.456 5.139 -41.41 6.570e-155 2.769e-151 343.6
STAB2 -1.917 4.889 -38.03 4.833e-142 1.528e-138 314.1
FCN3 -3.361 7.373 -36.66 1.153e-136 2.916e-133 301.7
CYP1A2 -4.718 7.868 -35.47 6.700e-132 1.412e-128 290.8
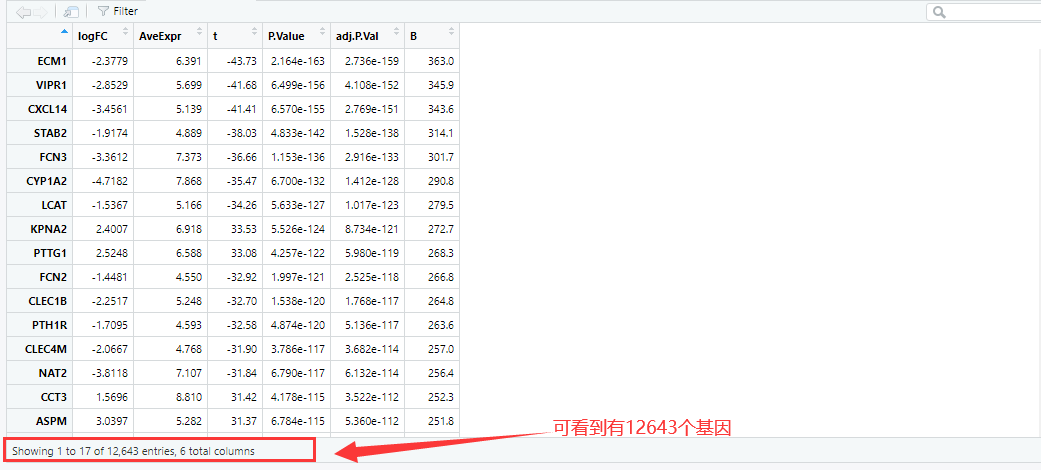
如果要提取某一特定基因,相关操作如下:
> tempOutput["CCL5",]
logFC AveExpr t P.Value adj.P.Val B
CCL5 -0.7769 5.547 -8.898 1.439e-17 5.193e-17 28.59
> tempOutput["VIPR1",]
logFC AveExpr t P.Value adj.P.Val B
VIPR1 -2.853 5.699 -41.68 6.499e-156 4.108e-152 345.9
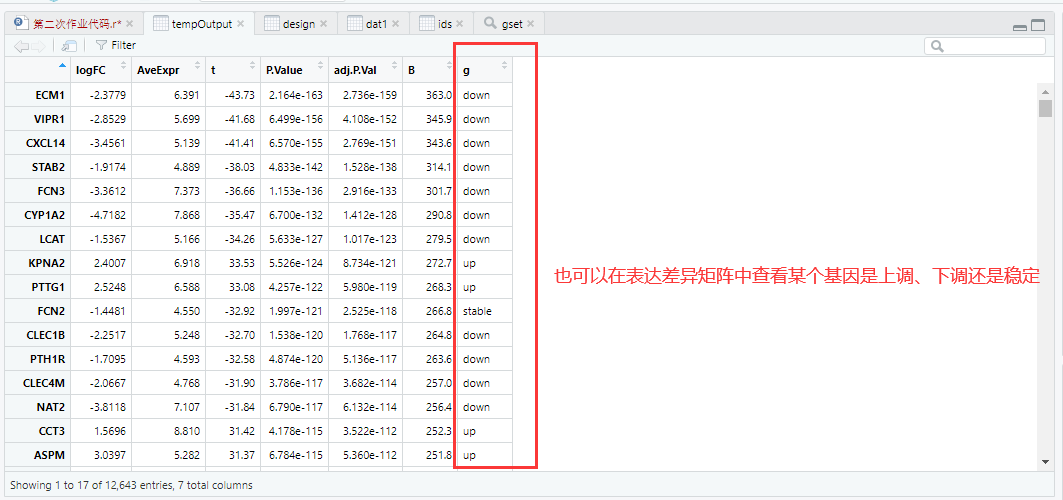
2.5.1.3、查看上调基因和下调基因
##上调基因和下调基因:使用了条件语句
tempOutput$g=ifelse(tempOutput$P.Value>0.01,'stable',
##if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( tempOutput$logFC >1.5,'up',
##接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( tempOutput$logFC < -1.5,'down','stable') )
##接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
> table(tempOutput$g)
down stable up
232 12325 86
可看到上调基因86个,下调基因232个,stable基因12325个。
2.5.2、该部分整体脚本展示
###四、基因的差异性分析
#####################################################
rm(list = ls()) ##一键清空
options(stringsAsFactors = F)
load(file = 'Expreset3.Rdata')##注意此处需要用探针ID转换为基于ID后的数据
dat1[1:4,1:4]##每次都要检测数据:可以看到行名为基因名
table(group_list1) ##table函数,查看group_list1中的分组个数
boxplot(dat1[1,]~group_list1) ##一个基因以group_list分组画箱线图
t.test(dat1[1,]~group_list1) ##对第一个基因做t检验
boxplot(dat1[2,]~group_list1) ##第二个基因以group_list分组画箱线图
t.test(dat1[2,]~group_list1) ##对第二个基因做t检验
bp=function(g){ ##自定义一个函数bp,函数为{}里的内容
library(ggpubr)
df=data.frame(gene=g,stage=group_list1)
p <- ggboxplot(df, x = "stage", y = "gene",
color = "stage", palette = "jco",
add = "jitter")
# Add p-value
p + stat_compare_means()
}
bp(dat1[3,]) ## 调用上面定义好的函数,避免同样的绘图代码重复多次敲。
##同理可尝试其它:
bp(dat1[1,])
bp(dat1[2,])
bp(dat1[4,])
##……
##差异分析走标准的limma流程
library(limma)
##创建一个分组的矩阵
design=model.matrix(~0+factor(group_list1))##创建一个分组的矩阵
colnames(design)=levels(factor(group_list1))
rownames(design)=colnames(dat1)
head(design)
##创建差异比较矩阵
contrast.matrix<-makeContrasts(paste0(unique(group_list1),collapse = "-"),levels = design)
contrast.matrix ##这个矩阵声明,我们要Tumor组和Non_Tumor组进行差异分析比较
##limma流程三板斧:
##第一步lmFit
##lmFit为每个基因给定一系列的阵列来拟合线性模型
fit<-lmFit(dat1,design)
##第二步eBayes
##eBayes给出了一个微阵列线性模型拟合,
##通过经验贝叶斯调整标准误差到一个共同的值来计算修正后的t统计量、修正后的f统计量和微分表达式的对数概率。
fit2<-contrasts.fit(fit, contrast.matrix)
fit2<-eBayes(fit2)
##第三步topTable
##topTable是从线性模型拟合中提取出排名靠前的基因表。
options(digits = 4) #设置全局的数字有效位数为4
##topTable(fit2,coef=2,adjust='BH')
##再运行以下我们输出的数据,就得到了相关基因
tempOutput<-topTable(fit2,coef=1,n=Inf)
tempOutput<-na.omit(tempOutput)##如果有NA值,则需要移除
head(tempOutput)
##提取其中特定基因:
tempOutput["CCL5",]
tempOutput["VIPR1",]
##上调基因和下调基因:使用了条件语句
tempOutput$g=ifelse(tempOutput$P.Value>0.01,'stable',
##if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( tempOutput$logFC >1.5,'up',
##接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( tempOutput$logFC < -1.5,'down','stable') )
##接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
table(tempOutput$g)
save(dat1,group_list1,tempOutput,file = 'tempOutput.Rdata')
#####################################################
2.6、基于上述步骤,绘制火山图、热图
2.6.1、分阶段解析演示
2.6.1.1、火山图
①数据加载:
rm(list = ls()) ## 一键清空
options(stringsAsFactors = F)
load(file = 'tempOutput.Rdata')
②以R自带函数绘制火山图
> DEG<-tempOutput##创建一个名为DEG的对象:后续对使用该数据进行增删查改操作,不影响原始数据
> head(DEG)
logFC AveExpr t P.Value adj.P.Val B g
ECM1 -2.378 6.391 -43.73 2.164e-163 2.736e-159 363.0 down
VIPR1 -2.853 5.699 -41.68 6.499e-156 4.108e-152 345.9 down
CXCL14 -3.456 5.139 -41.41 6.570e-155 2.769e-151 343.6 down
STAB2 -1.917 4.889 -38.03 4.833e-142 1.528e-138 314.1 down
FCN3 -3.361 7.373 -36.66 1.153e-136 2.916e-133 301.7 down
CYP1A2 -4.718 7.868 -35.47 6.700e-132 1.412e-128 290.8 down
attach(DEG)
plot(logFC,-log10(P.Value))##使用R语言自带的函数绘制火山图

③以ggpubr绘制火山图
> library(ggpubr)##使用ggpubr绘制火山图
> df=DEG##再创建一个新的对象,用于后续绘图的临时操作
> df$v= -log10(P.Value) ##在df中新增加一列'v',值为-log10(P.Value)
Ⅰ、基础绘制
> ggscatter(df, x = "logFC", y = "v",size=0.5) ##基础绘制

Ⅱ、美化处理(一)
##根据基因上调下调添加颜色ggscatter(df, x = "AveExpr", y = "logFC",size = 0.2)
ggscatter(df, x = "logFC", y = "v",size=0.5,color = 'g')

Ⅲ、美化处理(二)
改变x、y轴的初始图和添加颜色后的图
ggscatter(df, x = "AveExpr", y = "logFC",size = 0.2)

> df$p_c = ifelse(df$P.Value<0.001,'p<0.001',
+ ifelse(df$P.Value<0.01,'0.001<p<0.01','p>0.01'))
> table(df$p_c )
0.001<p<0.01 p<0.001 p>0.01
905 8561 3177
>ggscatter(df,x = "AveExpr", y = "logFC", color = "p_c",size=0.2,
palette = c("green", "red", "black") )

2.6.1.2、热图
①数据加载:
rm(list = ls()) ## 一键清空
options(stringsAsFactors = F)
load(file = 'tempOutput.Rdata')
②数据检测和基因表达情况
> ##每次都要检测数据
> dat1[1:4,1:4]
GSM362958 GSM362959 GSM362960 GSM362961
DDR1 5.494 6.311 6.564 5.060
RFC2 4.768 4.440 4.328 4.059
HSPA6 8.084 4.098 3.713 4.338
PAX8 3.737 3.921 3.876 4.301
> table(group_list1)
group_list1
Non_Tumor Tumor
220 225
③挑选差异最大、最小的100个基因
> ##挑选出差异最大100个基因和差异最小的100个基因
> x=tempOutput$logFC ##deg取logFC这列并将其重新赋值给x
> names(x)=row.names(tempOutput)##将基因名作为名字,命名给给x
> x[1:4] ##查看结果
ECM1 VIPR1 CXCL14 STAB2
-2.378 -2.853 -3.456 -1.917
cg=c(names(head(sort(x),100)),names(tail(sort(x),100)))
##对x进行从小到大排列,取前100及后100,并取其对应的基因名,作为向量赋值给cg
④绘制热图:下载与加载相关R包
> install.packages("pheatmap")
trying URL 'https://cran.rstudio.com/bin/windows/contrib/4.2/pheatmap_1.0.12.zip'
Content type 'application/zip' length 78905 bytes (77 KB)
downloaded 77 KB
程序包‘pheatmap’打开成功,MD5和检查也通过
下载的二进制程序包在
C:\Users\无咎\AppData\Local\Temp\Rtmpe0tV9z\downloaded_packages里
> library(pheatmap)##热图相关R包
Ⅰ、对dat按照cg取行,所得到的矩阵来画热图
> pheatmap(dat1[cg,],show_colnames =F,show_rownames = F)

Ⅱ、对log-ratio数值进行归一化后进行热图绘制
> n=t(scale(t(dat1[cg,])))##通过“scale”对log-ratio数值进行归一化
##scale()函数需要行是样本名,列是基因名,所以需要转置,然后再转置回来。
> n[n>2]=2
> n[n< -2]= -2
> n[1:4,1:4]
GSM362958 GSM362959 GSM362960 GSM362961
HAMP -1.692 -0.4746 -0.5258 0.93232
CYP1A2 -1.479 -0.9950 -1.0875 0.55476
MT1M -1.347 -0.6389 -1.3781 -0.08755
SLC22A1 -1.447 -1.5096 -1.0158 0.72860
> pheatmap(n,show_colnames =F,show_rownames = F)

Ⅲ、以‘Non_Tumor’和‘Tumor’分组画热图
ac=data.frame(g=group_list1)
> rownames(ac)=colnames(n) ##将ac的行名也就分组信息(是‘Non_Tumor’还是‘Tumor’)给到n的列名,即热图中位于上方的分组信息
> pheatmap(n,show_colnames =F,
+ show_rownames = F,
+ cluster_cols = F,
+ annotation_col=ac) ##列名注释信息为ac即分组信息

2.6.2、该部分整体脚本展示
## for volcano 火山图
#####################################################
rm(list = ls()) ## 一键清空
options(stringsAsFactors = F)
load(file = 'tempOutput.Rdata')
DEG<-tempOutput##创建一个名为DEG的对象:后续对使用该数据进行增删查改操作,不影响原始数据
head(DEG)
attach(DEG)
plot(logFC,-log10(P.Value))##使用R语言自带的函数绘制火山图
library(ggpubr)##使用ggpubr绘制火山图
df=DEG##再创建一个新的对象,用于后续绘图的临时操作
df$v= -log10(P.Value) ##在df中新增加一列'v',值为-log10(P.Value)
ggscatter(df, x = "logFC", y = "v",size=0.5) ##基础绘制
ggscatter(df, x = "logFC", y = "v",size=0.5,color = 'g')##根据基因上调下调添加颜色
ggscatter(df, x = "AveExpr", y = "logFC",size = 0.2)
df$p_c = ifelse(df$P.Value<0.001,'p<0.001',
ifelse(df$P.Value<0.01,'0.001<p<0.01','p>0.01'))
table(df$p_c )
ggscatter(df,x = "AveExpr", y = "logFC", color = "p_c",size=0.2,
palette = c("green", "red", "black") )
#####################################################
## for heatmap 热图
#####################################################
rm(list = ls()) ##一键清空
options(stringsAsFactors = F)
load(file = 'tempOutput.Rdata')
##每次都要检测数据
dat1[1:4,1:4]
table(group_list1)
##挑选出差异最大100个基因和差异最小的100个基因
x=tempOutput$logFC ##取logFC这列并将其重新赋值给x
names(x)=row.names(tempOutput)##将基因名作为名字,命名给给x
x[1:4]##查看结果
cg=c(names(head(sort(x),100)),names(tail(sort(x),100)))
##对x进行从小到大排列,取前100及后100,并取其对应的基因名,作为向量赋值给cg
library(pheatmap)##热图相关R包
pheatmap(dat1[cg,],show_colnames =F,show_rownames = F) ##对dat按照cg取行,所得到的矩阵来画热图
n=t(scale(t(dat1[cg,])))##通过“scale”对log-ratio数值进行归一化
##scale()函数需要行是样本名,列是基因名,所以我们需要转置,然后再转置回来。
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
pheatmap(n,show_colnames =F,show_rownames = F)
##以‘Non_Tumor’和‘Tumor’分组画热图
ac=data.frame(g=group_list1)
rownames(ac)=colnames(n)
##将ac的行名也就分组信息(是‘Non_Tumor’还是‘Tumor’)给到n的列名,即热图中位于上方的分组信息
pheatmap(n,show_colnames =F,
show_rownames = F,
cluster_cols = F,
annotation_col=ac) ##列名注释信息为ac即分组信息
#####################################################
相关参考:
1、R语言进行GEO数据挖掘与分析
2、主成分分析(PCA)原理详解
3、基于R语言做层次聚类分析
4、一个GSE有两个GPL该怎么办