github: 25280841/PhaVa: Adapting the phasefinder approach for identifying phase variation to long reads (github.com)
挺简单的,这里就不翻译了,大家看着直接用吧。
PhaVa
PhaVa is an approach for finding potentially Phase Variable invertible regions, also referred to as invertons, in long-read seqeuncing data
Dependencies
Versions listed are the versions PhaVa has been tested on.
- EMBOSS (v. 6.5.7) einverted
- minimap2 (v. 2.17)
- pysam (v. 0.17.0)
- Biopython (v. 1.81)
PhaVa is developed and tested on Linux operating systems (CentOS Linux 7), however it should compatible with Mac OSX and Windows
Usage
The PhaVa workflow is divided into three steps: locate, create, and ratio.
phava locate -i genome.fasta -d out_dir
phava create -d out_dir
phava ratio -r long_reads.fastq -d out_dir
Alternatively, all three steps can be run in a single command via variation_wf
phava variation_wf -i genome.fasta -r long_reads.fastq -d out_dir
Output from each step is centered around a output directory (-d) and should be the same directory for the entire workflow The locate and create steps only need to be performed once for a given genome or metagenome, and ratio can then be run on long-read samples using the same output directory (-d)
Any invertons with at least 1 read aligning in the reverse orientation will be found in the output. However, it is strongly recommended to fruther filter based on a minimum reverse read count and minimum % reverse of all reads cutoff (3 and 3% are recommended, respectively)
Expected output: 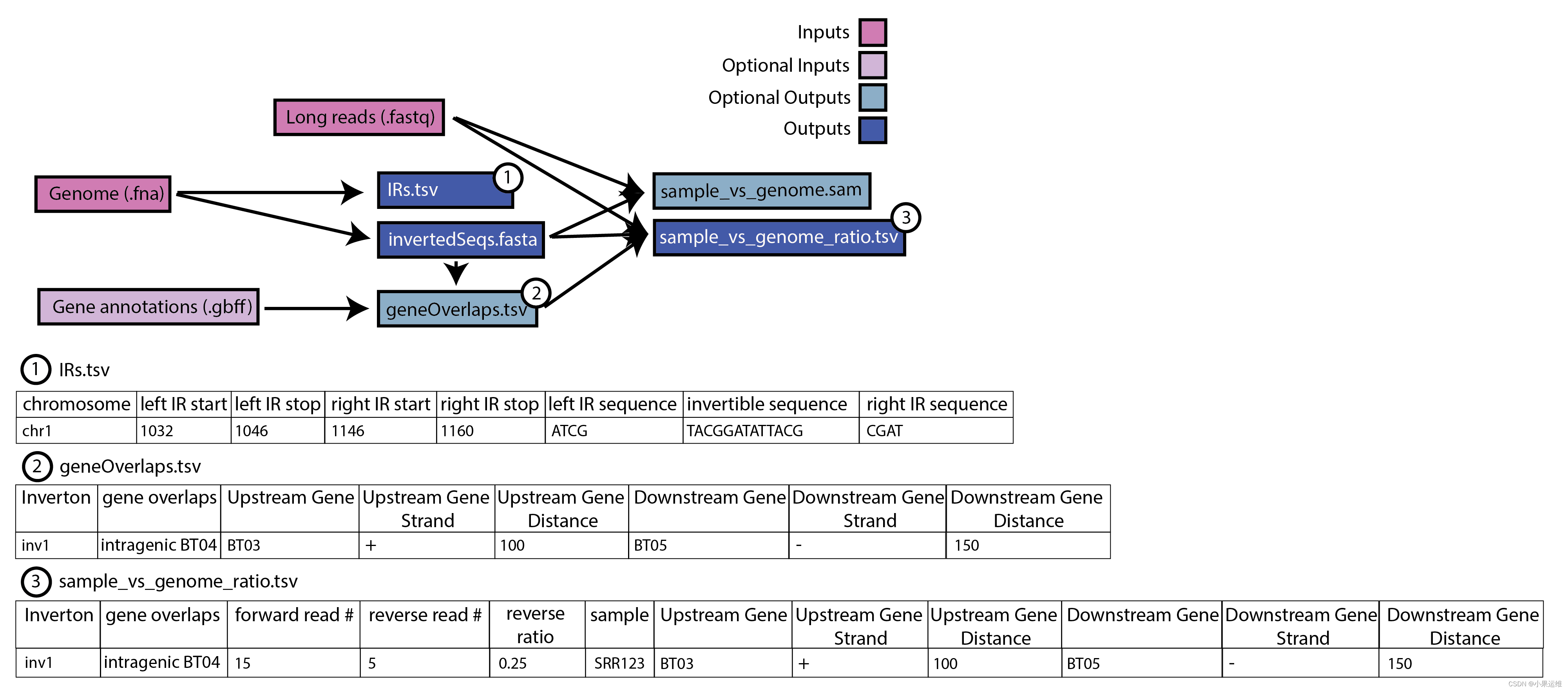
Installation
Beyond installing dependencies, PhaVa install is:
git clone https://github.com/patrickwest/PhaVa
Testing
PhaVa install can be tested on a small simulated dataset, typically in <1 minute, with pytest and a pytest module located in the 'tests' subdirectory:
pytest phava_test.py