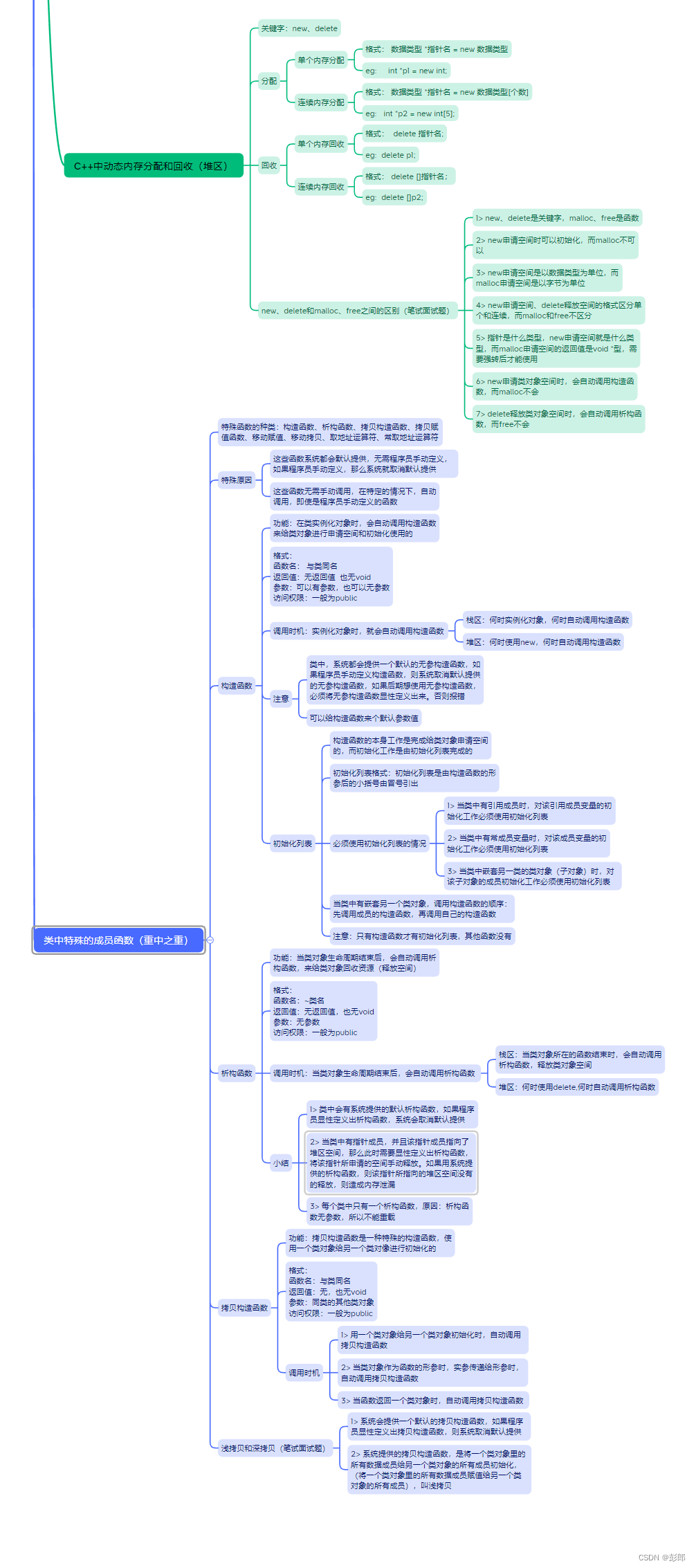
啥也不说了,这个好用,给大家推荐:YongxinLiu/EasyMicrobiome (github.com)
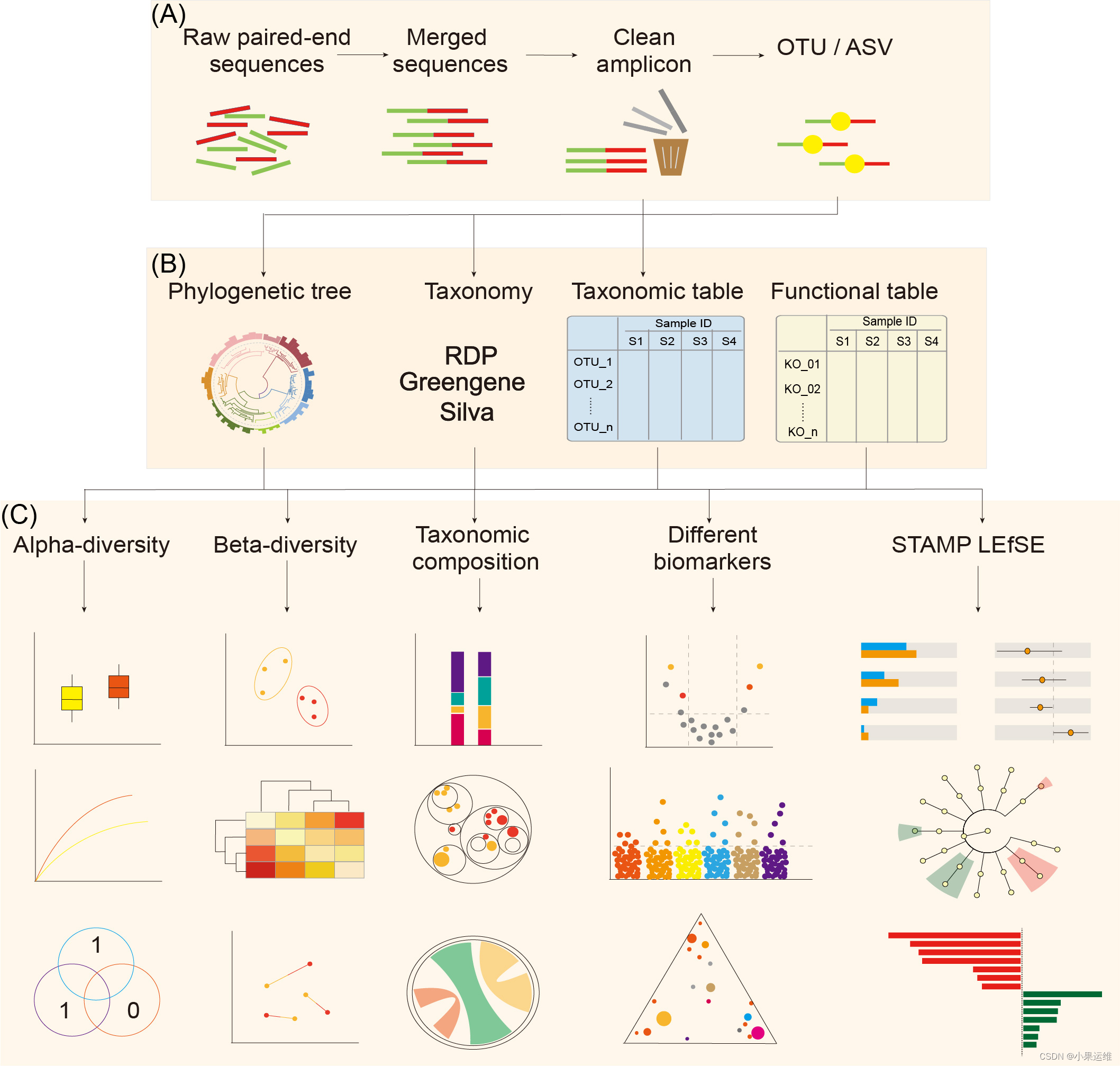
大家先看看引用文献吧,很有用:https://doi.org/10.1002/imt2.83
还有这个,后面马上介绍:YongxinLiu/EasyAmplicon: Easy Amplicon data analysis pipeline (github.com)
这个EasyMicrobiome的代码库包括了大家常用的一些脚本和数据库文件,大部分不需要任何依赖,唯一需要注意其中的R、python等脚本的版本,数据库文件可能不是最新的,大家到对应链接下载替换即可。
1、下载方法:
设置了很多途径,大家访问任何一个方式都可以下载:
https://github.com/yongxinliu/EasyMicrobiome
http://nmdc.cn/datadownload
百度网盘 请输入提取码
# 方法1. git下载,可使用wget或主页中直接下载压缩包
git clone https://github.com/YongxinLiu/EasyMicrobiome
# 方法2. 备用链接下载
wget -c ftp://download.nmdc.cn/tools/soft/EasyMicrobiome.tar.gz
tar xvzf EasyMicrobiome.tar.gz下面权限修改等就不说了吧,一般都会用,大家参考就行。
chmod +x EasyMicrobiome/linux/*
# 临时添加环境变量
export PATH=$PATH:`pwd`/EasyMicrobiome/linux:`pwd`/EasyMicrobiome/script"
# 将变量写入.bashrc,永久添加环境变量
echo "PATH=$PATH:`pwd`/EasyMicrobiome/linux:`pwd`/EasyMicrobiome/script" >> ~/.bashrc里面脚本大部分不需要安装,加入系统环境路径即可,但需要注意脚本需要的环境依赖
2、主要内容说明
主要软件清单
名称的链接对应软件的主页,大部分已经整合入本项目。对于较大的文件,标题后提供下载链接,使用时需自行下载。
- linux:Linux系统下分析软件
- microbiome_helper:微生物组分析输助脚本,如metaphlan2结果转换STAMP格式(metaphlan_to_stamp.pl),picurst结果功能组成绘图(plot_metagenome_contributions.R)
- Miniconda3-latest-Linux-x86_64.sh:软件管理器 https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
- qiime2-2023.2.tar.gz:QIIME2安装包,解压至conda的envs目录可用 ftp://download.nmdc.cn/tools/conda/qiime2-2023.2.tar.gz
- qiime2-2023.2-py38-linux-conda.yml:QIIME2软件安装清单,使用conda在线安装
- sparcc:sparcc网络分析python脚本
- usearch:扩增子分析流程
- vsearch:扩增子分析流程(免费64位版usearch)
- mac:Mac系统下分析软件
- csvtk:表格分析工具
- iqtree:进化树构建
- qiime2-2023.9-py38-osx-conda.yml:QIIME2软件安装清单,使用conda在线安装
- R-4.3.2.pkg:R语言安装包
- RStudio-2023.03.0-386.dmg:RStudio安装包
- rush:并行管理工具
- seqkit:序列处理工具
- taxonkit:NCBI分类处理工具
- usearch:扩增子分析流程
- vsearch:扩增子分析流程(免费64位版usearch)
- win:Windows系统下分析软件
- Git-2.40.0-64-bit.exe:提供Git bash环境,自行下载安装,教程见:Windows轻松实现linux shell环境:gitforwindows
- R-4.3.2-win.exe:R语言安装包,下载最新版:Downad CRAN - China Tsinghua - Download R for Windows(Mac) —— base —— Download R 4.3.2
- RStudio-2023.03.0-386.exe:RStudio安装包,提供分析运行界面。
- 4.2.zip:R语言常用400+包合集,解压至R包安装位置即可用。
- usearch.exe:扩增子分析流程
- vsearch.exe:扩增子分析流程(免费64位版usearch)
- STAMP2.1.3:微生物组图形界面差异分析工具
- Adobe_Illustrator_CC_2018_v22.1.0.314_x64_zh_CN_Portable.7z:图片拼图、模式图绘制工具,使用试用版或自行购买
- Cytoscape_3_8_2_windows_64bit.exe:网络分析安装包
- csvtk.exe:表格分析工具
- seqkit.exe:序列处理工具
- taxonkit.exe:NCBI分类处理工具
- rush.exe:并行管理工具
- epp510_1828_64bit.exe:文本编辑器
- Xshell:远程访问服务器终端,需要申请免费版下载链接;备选PuTTY
- FileZilla:远程访问服务器文件上传下载,备选WinSCP
- gephi-0.9.2-windows.exe:网络图绘制工具
- iqtree.exe:进化树构建
- libiomp5md.dll:动态库,iqtree运行中提示缺少时,可添加至软件所在目录
- jdk-11.0.7_windows-x64_bin.exe:Java运行环境
- muscle.exe:多序列比对工具
- npp.7.8.9.Installer.x64.exe:文本编辑器NotePad++安装包
- rtools40-x86_64.exe:R源码安装时的编绎工具
- wget.exe:命令行下载工具
包含的数据库
- gg:GreenGenes细菌16S数据库
- gg_13_8_otus.tar.gz:13年8月更新OTU数据库,用于usearch有参定量和PICRUSt/BugBase功能预测、QIIME 2制作分类器。国内备份链接
- 16S_13_5_precalculated.tab.gz:picrust的GreenGenes 16S拷贝数
- ko_13_5_precalculated.tab.gz:picrust的GreenGenes 16S对应的KO数量信息
- kegg:KEGG数据库描述信息整理
- ko00001.keg:KEGG层级注释体系,主页 —— KEGG BRITE —— KEGG Orthology (KO) —— Download htext,下载保存为ko00001.tsv
- ko00001.tsv:转换jason格式为制表符分隔的KO对应描述、(三级)通路、二级通路和一级通路信息
- KO1-4.txt:KO对应的3级注释,包括(三级)通路、二级通路和一级通路信息,用于KO表的分类汇总
- KO_description.txt:KO编号对应的功能描述
- KO_path.list:KO与通路(Pathway)的对应关系,存在某个KO存在于多个通路(1对多)
- usearch:usearch/vsearch物种分类sintax命令使用数据库
- rdp_16s_v18.fa.gz:16S的RDP16数据库,usearch作者整理,更多16S、ITS和18S数据库见 SINTAX downloads
- rdp_16s_v18.fa.gz:16S的RDP18数据库,2021年基于RDP数据库整理
- utax_reference_dataset_all_04.02.2020.fasta.gz:ITS注释数据库,可从UNITE下载
- eggnog: eggnog结果的注释文件补充
- COG.anno:COG的第一、二级注释
脚本
-
使用说明:分析常用脚本类型
- .R文件为R脚本,使用Rscript命令执行;
- .sh为Shell脚本,使用/bin/bash命令执行;
- .pl为Perl脚本,使用perl命令执行;
- .py为Python脚本,使用python执行,注意还分为python2和python3两种
-
script:微生物组数据分析
- BugBase:16S扩增子表型预测R脚本和数据库
- FAPROTAX_1.2.4:16S扩增子元素循环预测Python脚本和数据库
- table2itol:iTOL进化树注释文件制作R脚本
- alpha_barplot.R:Alpha多样性指数柱状图+标准差图绘制
- alpha_boxplot.R:Alpha多样性指数箱线图+统计绘制
- alpha_rare_curve.R:usearch计算稀释曲线可视化
- beta_cpcoa.R:基于距离矩阵开展限制性PCoA分析及可视化散点图+分组着色+置信椭圆,要求至少3个分组
- beta_pcoa.R:基于距离矩阵的主坐标PCoA分析及可视化散点图+分组着色+置信椭圆+组间两两统计
- BetaDiv.R:更多Beta多样性分析,如PCA、PCoA、NMDS、LDA、CCA、RDA等
- compare.R:两组比较,支持t.test、wilcox、edgeR三种方法
- compare_heatmap.R/sh:基于两组比较结果绘制热图
- compare_manhattan.sh:基于两组比较结果绘制曼哈顿图
- compare_volcano.R:基于两组比较结果绘制火山图
- faprotax_report_sum.pl:FARPROTAX分析结果报告整理
- filter_feature_table.R:按频率过滤OTU表
- format_dbcan2list.pl:dbcan数据库注释结果整理
- format2lefse.R:OTU表和物种注释生成LEfSe输入文件
- format2stamp.R:OTU表和物种注释生成STAMP输入文件
- kegg_ko00001_htext2tsv.pl:KEGG注释结果整理
- kraken2alpha.R:Kraken2结果整理、抽平和alpha多样性指数计算
- mat_gene2ko.R:按类型折叠表格
- metaphlan_boxplot.R:metaphalan2结果可视化为箱线图
- metaphlan_hclust_heatmap.R:metaphalan2结果可视化为聚类热图
- metaphlan_to_stamp.pl:metaphalan2结果转换为STAMP格式
- otu_mean.R:OTU表统计分组均值(总体均值)、分组求合
- otutab_filter_nonBac.R:16S的OTU表按sintax注释结果选择细菌、古菌且过滤叶绿体和线粒体
- otutab_filter_nonFungi.R:ITS的OTU表选择真菌
- otutab_freq2count.R:转换频率为伪整数,用于要求整型输入的分析,如多样性、edgeR差异分析等
- otutab_rare.R:OTU表抽平
- plot_metagenome_contributions.R:PICRUSt结果物种的功能组成绘制
- sp_pheatmap.sh:绘制热图
- sp_vennDiagram.sh:绘制维恩图
- summarizeAbundance.py:按类型折叠大表,如基因按KEGG的KO合并
- tax_circlize.R:物种组成圈图
- tax_maptree.R:物种组成气泡图
- tax_stackplot.R:物种组成堆叠柱状图
3、脚本介绍及使用示例:
这里面的脚本最大的好处就是每个脚本里面都已经将每个参数包括输入输出等都已经注释说明,大家可直接根据参数调整使用
当然最有用的地方还是在于能支撑扩增子分析流程和宏基因组分析流程,这个是刘永鑫提供的两个流程代码,做出的图也是相当漂亮:
GitHub - YongxinLiu/EasyAmplicon: Easy Amplicon data analysis pipelineEasy Amplicon data analysis pipeline . Contribute to YongxinLiu/EasyAmplicon development by creating an account on GitHub.![]() https://github.com/YongxinLiu/EasyAmplicon
https://github.com/YongxinLiu/EasyAmplicon
GitHub - YongxinLiu/EasyMetagenome: Easy Metagenome PipelineEasy Metagenome Pipeline. Contribute to YongxinLiu/EasyMetagenome development by creating an account on GitHub.![]() https://github.com/YongxinLiu/EasyMetagenome扩增子分析流程,分析内容和结果相当丰富
https://github.com/YongxinLiu/EasyMetagenome扩增子分析流程,分析内容和结果相当丰富
# 易扩增子EasyAmplicon
# 作者 Authors: 刘永鑫(Yong-Xin Liu), 陈同(Tong Chen)等
# 版本 Version: v1.20
# 更新 Update: 2023-10-13
# 系统要求 System requirement: Windows 10+ / Mac OS 10.12+ / Ubuntu 20.04+
# 引文 Reference: Liu, et al. 2023. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based
# pipeline for amplicon data analysis in microbiome research. iMeta 2: e83. https://doi.org/10.1002/imt2.83
# 设置工作(work directory, wd)和软件数据库(database, db)目录
# 添加环境变量,并进入工作目录 Add environmental variables and enter work directory
# **每次打开Rstudio必须运行下面4行 Run it**,可选替换${db}为EasyMicrobiome安装位置
wd=/d/amplicon
db=/d/EasyMicrobiome
PATH=$PATH:${db}/win
cd ${wd}
## 1. 起始文件 start files
# 1. 分析流程pipeline.sh
# 2. 样本元信息metadata.txt,保存于result目录
# 3. 测序数据fastq文件保存于seq目录,通常以`.fq.gz`结尾,每个样品一对文件
# 4. 创建临时文件存储目录,分析结束可删除
mkdir -p seq result temp
### 1.1. 元数据/实验设计 metadata
# 准备样本元数据result/metadata.txt
# csvtk统计表行(样本数,不含表头)列数,-t设置列分隔为制表符,默认为;
csvtk -t stat result/metadata_raw.txt
# 元数据至少3列,首列为样本ID(SampleID),结尾列为描述(Description)
# cat查看文件,-A显示符号,"|"为管道符实现命令连用,head显示文件头,-n3控制范围前3行
cat -A result/metadata_raw.txt | head -n3
# windows用户结尾有^M,运行sed命令去除,再用cat -A检查
sed 's/\r//' result/metadata_raw.txt > result/metadata.txt
cat -A result/metadata.txt | head -n3
### 1.2. 测序数据 sequencing data
# # 本段代码可在RStudio中Ctrl + Shift + C 取消注释“#”后运行
# # (可选)下载测序数据,按GSA的CRA(批次)和CRR(样品)编号下载数据
# # 示例下载单个文件并改名
# mkdir -p seq
# wget -c ftp://download.big.ac.cn/gsa/CRA002352/CRR117575/CRR117575_f1.fq.gz -O seq/KO1_1.fq.gz
# # 按实验设计编号批量下载并改名
# awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_f1.fq.gz -O seq/"$1"_1.fq.gz")}' \
# <(tail -n+2 result/metadata.txt)
# awk '{system("wget -c ftp://download.big.ac.cn/gsa/"$5"/"$6"/"$6"_r2.fq.gz -O seq/"$1"_2.fq.gz")}' \
# <(tail -n+2 result/metadata.txt)
# 公司返回的测序结果,通常为一个样品一对fq/fastq.gz格式压缩文件
# 文件名与样品名务必对应:不一致时手工修改,批量改名见"常见问题6"
# 如果测序数据是.gz的压缩文件,有时需要使用gunzip解压后使用,vsearch通常可直接读取压缩文件
# gunzip seq/*.gz
# zless按页查看压缩文件,空格翻页、q退出;head默认查看前10行,-n指定行
ls -sh seq/
zless seq/KO1_1.fq.gz | head -n4
# 每行太长,指定查看每行的1-60个字符
zless seq/KO1_1.fq | head | cut -c 1-60
# 统计测序数据,依赖seqkit程序
seqkit stat seq/KO1_1.fq.gz
# 批量统计测序数据并汇总表
seqkit stat seq/*.fq.gz > result/seqkit.txt
head result/seqkit.txt
### 1.3. 流程和数据库 pipeline & database
# 数据库第一次使用必须解压,以后可跳过此段
# usearchs可用16S/18S/ITS数据库:RDP, SILVA和UNITE,本地文件位置 ${db}/usearch/
# usearch数据库database下载页: http://www.drive5.com/usearch/manual/sintax_downloads.html
# 解压16S RDP数据库,gunzip解压缩,seqkit stat统计
# 保留原始压缩文件
gunzip -c ${db}/usearch/rdp_16s_v18.fa.gz > ${db}/usearch/rdp_16s_v18.fa
seqkit stat ${db}/usearch/rdp_16s_v18.fa # 2.1万条序列
# 解压ITS UNITE数据库,需自行从官网或网盘db/amplicon/usearch中下载
# gunzip -c ${db}/usearch/utax_reference_dataset_all_25.07.2023.fasta.gz >${db}/usearch/unite.fa
seqkit stat ${db}/usearch/unite.fa # 32.6万
# Greengene数据库用于功能注释: ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz
# 默认解压会删除原文件,-c指定输出至屏幕,> 写入新文件(可改名)
gunzip -c ${db}/gg/97_otus.fasta.gz > ${db}/gg/97_otus.fa
seqkit stat ${db}/gg/97_otus.fa
## 2. 序列合并和重命名 reads merge and rename
### 2.1 合并双端序列并按样品重命名 Merge pair-end reads and rename
#测试.以WT1单样品合并为例
#time统计计算时间,real是物理时间,user是计算时间,sys是硬件等待时间
# time vsearch --fastq_mergepairs seq/WT1_1.fq.gz \
# --reverse seq/WT1_2.fq.gz \
# --fastqout temp/WT1.merged.fq \
# --relabel WT1.
# head temp/WT1.merged.fq
#依照实验设计批处理并合并
#tail -n+2去表头,cut -f1取第一列,获得样本列表;18个样本x1.5万对序列合并8s
#Win下复制Ctrl+C为Linux下中止,为防止异常中断,结尾添加&转后台,无显示后按回车继续
# 一部分电脑 rush 不支持,运行时调度失败,请使用 for 循环部分
# for 循环部分是放入后台运行的,点完 run 之后,看上去程序已运行完,实际没运行完,而是正在运行中。
# 不要急于运行后面的程序。
# 之前课程,有发现每次运行结果都不一样,就是因为 for 循环部分没运行完,只生成了部分数据,导致后面
# 每个样品 reads 数每次运行都会不一致。
#方法1.for循环顺序处理
# time for i in `tail -n+2 result/metadata.txt|cut -f1`;do
# vsearch --fastq_mergepairs seq/${i}_1.fq.gz --reverse seq/${i}_2.fq.gz \
# --fastqout temp/${i}.merged.fq --relabel ${i}.
# done &
# 一部分电脑 rush 不支持,运行时调度失败,请使用 for 循环部分
#方法2.rush并行处理,任务数jobs(j),2可加速1倍4s;建议设置2-4
time tail -n+2 result/metadata.txt | cut -f 1 | \
rush -j 2 "vsearch --fastq_mergepairs seq/{}_1.fq.gz --reverse seq/{}_2.fq.gz \
--fastqout temp/{}.merged.fq --relabel {}."
# 检查最后一个文件前10行中样本名
head temp/`tail -n+2 result/metadata.txt | cut -f 1 | tail -n1`.merged.fq | grep ^@
##方法3.不支持压缩文件时解压再双端合并
# time tail -n+2 result/metadata.txt | cut -f 1 | \
# rush -j 1 "vsearch --fastq_mergepairs <(zcat seq/{}_1.fq.gz) --reverse <(zcat seq/{}_2.fq.gz) \
# --fastqout temp/{}.merged.fq --relabel {}."
#
# time for i in `tail -n+2 result/metadata.txt|cut -f1`;do
# vsearch --fastq_mergepairs <(zcat seq/${i}_1.fq.gz) --reverse <(zcat seq/${i}_2.fq.gz) \
# --fastqout temp/${i}.merged.fq --relabel ${i}.
# done &
### 2.2 (可选)单端文件改名 Single-end reads rename
# # 单个序列改名示例
# i=WT1
# gunzip -c seq/${i}_1.fq.gz > seq/${i}.fq
# usearch -fastx_relabel seq/${i}.fq -fastqout temp/${i}.merged.fq -prefix ${i}.
#
# # 批量改名,需要有单端fastq文件,且解压(usearch不支持压缩格式)
# gunzip seq/*.gz
# time for i in `tail -n+2 result/metadata.txt|cut -f1`;do
# usearch -fastx_relabel seq/${i}.fq -fastqout temp/${i}.merged.fq -prefix ${i}.
# done &
# # vsearch大数据方法参考“常见问题2”
### 2.3 改名后序列整合 integrate renamed reads
#合并所有样品至同一文件
cat temp/*.merged.fq > temp/all.fq
#查看文件大小223M,软件不同版本结果略有差异
ls -lsh temp/all.fq
# 查看序列名,“.”之前是否为样本名,样本名绝不允许有点 (".")
# 样本名有点 (.) 的一个显著特征是生成的特征表会很大,特征表里面列很多,导致后面分析出现内存不足。
# 后面分析获得特征表后要看一眼有没有问题,遇到内存不足问题,也要回头来排查。
head -n 6 temp/all.fq|cut -c1-60
## 3. 切除引物与质控 Cut primers and quality filter
# 左端10bp标签+19bp上游引物V5共为29,右端V7为18bp下游引物
# Cut barcode 10bp + V5 19bp in left and V7 18bp in right
# 务必清楚实验设计和引物长度,引物已经去除可填0,27万条序列14s
time vsearch --fastx_filter temp/all.fq \
--fastq_stripleft 29 --fastq_stripright 18 \
--fastq_maxee_rate 0.01 \
--fastaout temp/filtered.fa
# 查看文件了解fa文件格式
head temp/filtered.fa
## 4. 去冗余挑选OTU/ASV Dereplicate and cluster/denoise
### 4.1 序列去冗余 Dereplicate
# 并添加miniuniqusize最小为8或1/1M,去除低丰度噪音并增加计算速度
# -sizeout输出丰度, --relabel必须加序列前缀更规范, 1s
vsearch --derep_fulllength temp/filtered.fa \
--minuniquesize 10 --sizeout --relabel Uni_ \
--output temp/uniques.fa
#高丰度非冗余序列非常小(500K~5M较适合),名称后有size和频率
ls -lsh temp/uniques.fa
# Uni_1;size=6423 - 去冗余后序列的名字 Uni_1;该序列在所有样品测序数据中出现 6423 次
# 为出现最多的序列。
head -n 2 temp/uniques.fa
### 4.2 聚类OTU/去噪ASV Cluster or denoise
#有两种方法:推荐unoise3去噪获得单碱基精度ASV,备选传统的97%聚类OTU(属水平精度)
#usearch两种特征挑选方法均自带de novo去嵌合体
#-minsize二次过滤,控制OTU/ASV数量至1-5千,方便下游统计分析
#方法1. 97%聚类OTU,适合大数据/ASV规律不明显/reviewer要求
#结果耗时1s, 产生508 OTUs, 去除126 chimeras
# usearch -cluster_otus temp/uniques.fa -minsize 10 \
# -otus temp/otus.fa \
# -relabel OTU_
#方法2. ASV去噪 Denoise: predict biological sequences and filter chimeras
#6s, 1530 good, 41 chimeras, 序列百万条可能需要几天/几周
usearch -unoise3 temp/uniques.fa -minsize 10 \
-zotus temp/zotus.fa
#修改序列名:Zotu为改为ASV方便识别
sed 's/Zotu/ASV_/g' temp/zotus.fa > temp/otus.fa
head -n 2 temp/otus.fa
#方法3. 数据过大无法使用usearch时,备选vsearch方法见"常见问题3"
### 4.3 基于参考去嵌合 Reference-based chimera detect
# 不推荐,容易引起假阴性,因为参考数据库无丰度信息
# 而de novo时要求亲本丰度为嵌合体16倍以上防止假阴性
# 因为已知序列不会被去除,数据库选择越大越合理,假阴性率最低
mkdir -p result/raw
# 方法1. vsearch+rdp去嵌合(快但容易假阴性)
# 可自行下载silva并解压(替换rdp_16s_v18.fa为silva_16s_v123.fa),极慢但理论上更好
vsearch --uchime_ref temp/otus.fa \
-db ${db}/usearch/rdp_16s_v18.fa \
--nonchimeras result/raw/otus.fa
# RDP: 7s, 143 (9.3%) chimeras; SILVA:9m, 151 (8.4%) chimeras
# Win vsearch结果添加了windows换行符^M需删除,mac不要执行此命令
sed -i 's/\r//g' result/raw/otus.fa
# 方法2. 不去嵌合
# cp -f temp/otus.fa result/raw/otus.fa
## 5. 特征表构建和筛选 Feature table create and filter
# OTU和ASV统称为特征(Feature),它们的区别是:
# OTU通常按97%聚类后挑选最高丰度或中心的代表性序列;
# ASV是基于序列进行去噪(排除或校正错误序列,并挑选丰度较高的可信序列)作为代表性序列
### 5.1 生成特征表
# id(1):100%相似度比对49.45%序列,1m50s
# id(0.97):97%相似度比对83.66%序列,1m10s(更高数据使用率,更快)
time vsearch --usearch_global temp/filtered.fa \
--db result/raw/otus.fa \
--id 0.97 --threads 4 \
--otutabout result/raw/otutab.txt
#212862 of 268019 (79.42%)可比对
# vsearch结果windows用户删除换行符^M校正为标准Linux格式
sed -i 's/\r//' result/raw/otutab.txt
head -n6 result/raw/otutab.txt | cut -f 1-6 |cat -A
# csvtk统计表行列
# 这里一定看好列数,是不是等于你的样品数;如果不等,一般是样品命名存在问题,具体看上面解释
csvtk -t stat result/raw/otutab.txt
### 5.2 物种注释,且/或去除质体和非细菌 Remove plastid and non-Bacteria
# 物种注释-去除质体和非细菌/古菌并统计比例(可选)
# RDP物种注释(rdp_16s_v18)更快,但缺少完整真核来源数据,可能不完整,耗时15s;
# SILVA数据库(silva_16s_v123.fa)更好注释真核、质体序列,极慢耗时3h起
# 置信阈值通常0.6/0.8,vserch最低0.1/usearch可选0输出最相似物种注释用于观察潜在分类
vsearch --sintax result/raw/otus.fa \
--db ${db}/usearch/rdp_16s_v18.fa \
--sintax_cutoff 0.1 \
--tabbedout result/raw/otus.sintax
sed -i 's/\r//' result/raw/otus.sintax
# 方法1. 原始特征表行数
wc -l result/raw/otutab.txt
#R脚本选择细菌古菌(真核)、去除叶绿体、线粒体并统计比例;输出筛选并排序的OTU表
#输入为OTU表result/raw/otutab.txt和物种注释result/raw/otus.sintax
#输出筛选并排序的特征表result/otutab.txt和
#统计污染比例文件result/raw/otutab_nonBac.txt和过滤细节otus.sintax.discard
#真菌ITS数据,请改用otutab_filter_nonFungi.R脚本,只筛选真菌
# Rscript ${db}/script/otutab_filter_nonBac.R -h # 显示参数说明
Rscript ${db}/script/otutab_filter_nonBac.R \
--input result/raw/otutab.txt \
--taxonomy result/raw/otus.sintax \
--output result/otutab.txt\
--stat result/raw/otutab_nonBac.stat \
--discard result/raw/otus.sintax.discard
# 筛选后特征表行数
wc -l result/otutab.txt
#过滤特征表对应序列
cut -f 1 result/otutab.txt | tail -n+2 > result/otutab.id
usearch -fastx_getseqs result/raw/otus.fa \
-labels result/otutab.id -fastaout result/otus.fa
#过滤特征表对应序列注释
awk 'NR==FNR{a[$1]=$0}NR>FNR{print a[$1]}'\
result/raw/otus.sintax result/otutab.id \
> result/otus.sintax
# 方法2. 觉得筛选不合理可以不筛选
# cp result/raw/otu* result/
#可选统计方法:OTU表简单统计 Summary OTUs table
usearch -otutab_stats result/otutab.txt \
-output result/otutab.stat
cat result/otutab.stat
#注意最小值、分位数,或查看result/raw/otutab_nonBac.stat中样本详细数据量,用于重采样
### 5.3 等量抽样标准化
# Normlize by subsample
#使用vegan包进行等量重抽样,输入reads count格式Feature表result/otutab.txt
#可指定输入文件、抽样量和随机数,输出抽平表result/otutab_rare.txt和多样性alpha/vegan.txt
mkdir -p result/alpha
Rscript ${db}/script/otutab_rare.R --input result/otutab.txt \
--depth 10000 --seed 1 \
--normalize result/otutab_rare.txt \
--output result/alpha/vegan.txt
usearch -otutab_stats result/otutab_rare.txt \
-output result/otutab_rare.stat
cat result/otutab_rare.stat
## 6. α多样性 alpha diversity
### 6.1. 计算α多样性 calculate alpha diversity
# 使用USEARCH计算14种alpha多样性指数(Chao1有错勿用)
#details in http://www.drive5.com/usearch/manual/alpha_metrics.html
usearch -alpha_div result/otutab_rare.txt \
-output result/alpha/alpha.txt
### 6.2. 计算稀释丰富度 calculate rarefaction richness
#稀释曲线:取1%-100%的序列中OTUs数量,每次无放回抽样
#Rarefaction from 1%, 2% .. 100% in richness (observed OTUs)-method without_replacement https://drive5.com/usearch/manual/cmd_otutab_subsample.html
usearch -alpha_div_rare result/otutab_rare.txt \
-output result/alpha/alpha_rare.txt \
-method without_replacement
#预览结果
head -n2 result/alpha/alpha_rare.txt
#样本测序量低出现非数值"-"的处理,详见常见问题8
sed -i "s/-/\t0.0/g" result/alpha/alpha_rare.txt
### 6.3. 筛选高丰度菌 Filter by abundance
#计算各特征的均值,有组再求分组均值,需根据实验设计metadata.txt修改组列名
#输入文件为feautre表result/otutab.txt,实验设计metadata.txt
#输出为特征表按组的均值-一个实验可能有多种分组方式
#-h显示脚本帮助(参数说明)
Rscript ${db}/script/otu_mean.R -h
#scale是否标准化,zoom标准化总和,all输出全部样本均值,type计算类型mean或sum
Rscript ${db}/script/otu_mean.R --input result/otutab.txt \
--metadata result/metadata.txt \
--group Group --thre 0 \
--scale TRUE --zoom 100 --all TRUE --type mean \
--output result/otutab_mean.txt
# 结果为全部和各组均值
head -n3 result/otutab_mean.txt
#如以平均丰度>0.1%筛选,可选0.5或0.05,得到每个组的OTU组合
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {for(i=3;i<=NF;i++) a[i]=$i; print "OTU","Group";} \
else {for(i=3;i<=NF;i++) if($i>0.1) print $1, a[i];}}' \
result/otutab_mean.txt > result/alpha/otu_group_exist.txt
head result/alpha/otu_group_exist.txt
cut -f 2 result/alpha/otu_group_exist.txt | sort | uniq -c
# 试一试:不同丰度下各组有多少OTU/ASV
# 可在 http://ehbio.com/test/venn/ 中绘图并显示各组共有和特有维恩或网络图
# 也可在 http://www.ehbio.com/ImageGP 绘制Venn、upSetView和Sanky
## 7. β多样性 Beta diversity
#结果有多个文件,需要目录
mkdir -p result/beta/
#基于OTU构建进化树 Make OTU tree, 4s
usearch -cluster_agg result/otus.fa -treeout result/otus.tree
#生成5种距离矩阵:bray_curtis, euclidean, jaccard, manhatten, unifrac
usearch -beta_div result/otutab_rare.txt -tree result/otus.tree \
-filename_prefix result/beta/
## 8. 物种注释分类汇总
#OTU对应物种注释2列格式:去除sintax中置信值,只保留物种注释,替换:为_,删除引号
cut -f 1,4 result/otus.sintax \
|sed 's/\td/\tk/;s/:/__/g;s/,/;/g;s/"//g' \
> result/taxonomy2.txt
head -n3 result/taxonomy2.txt
#OTU对应物种8列格式:注意注释是非整齐
#生成物种表格OTU/ASV中空白补齐为Unassigned
awk 'BEGIN{OFS=FS="\t"}{delete a; a["k"]="Unassigned";a["p"]="Unassigned";a["c"]="Unassigned";a["o"]="Unassigned";a["f"]="Unassigned";a["g"]="Unassigned";a["s"]="Unassigned";\
split($2,x,";");for(i in x){split(x[i],b,"__");a[b[1]]=b[2];} \
print $1,a["k"],a["p"],a["c"],a["o"],a["f"],a["g"],a["s"];}' \
result/taxonomy2.txt > temp/otus.tax
sed 's/;/\t/g;s/.__//g;' temp/otus.tax|cut -f 1-8 | \
sed '1 s/^/OTUID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' \
> result/taxonomy.txt
head -n3 result/taxonomy.txt
#统计门纲目科属,使用 rank参数 p c o f g,为phylum, class, order, family, genus缩写
mkdir -p result/tax
for i in p c o f g;do
usearch -sintax_summary result/otus.sintax \
-otutabin result/otutab_rare.txt -rank ${i} \
-output result/tax/sum_${i}.txt
done
sed -i 's/(//g;s/)//g;s/\"//g;s/\#//g;s/\/Chloroplast//g' result/tax/sum_*.txt
# 列出所有文件
wc -l result/tax/sum_*.txt
head -n3 result/tax/sum_g.txt
## 9. 有参定量特征表
# 比对Greengenes97% OTUs比对,用于PICRUSt/Bugbase功能预测
mkdir -p result/gg/
# usearch比对更快,但文件超限报错选附录14 vsearch比对
# 默认10核以下使用1核,10核以上使用10核
usearch -otutab temp/filtered.fa -otus ${db}/gg/97_otus.fa \
-otutabout result/gg/otutab.txt -threads 4
# 比对率80.0%, 1核11m,4核3m,10核2m,内存使用743Mb
head -n3 result/gg/otutab.txt
#统计
usearch -otutab_stats result/gg/otutab.txt -output result/gg/otutab.stat
cat result/gg/otutab.stat
## 10. 空间清理及数据提交
#删除中间大文件
rm -rf temp/*.fq
# 分双端统计md5值,用于数据提交
cd seq
md5sum *_1.fq.gz > md5sum1.txt
md5sum *_2.fq.gz > md5sum2.txt
paste md5sum1.txt md5sum2.txt | awk '{print $2"\t"$1"\t"$4"\t"$3}' | sed 's/*//g' > ../result/md5sum.txt
rm md5sum*
cd ..
cat result/md5sum.txt
# R语言多样性和物种组成分析
## 1. Alpha多样性
### 1.1 Alpha多样性箱线图
# 查看帮助
Rscript ${db}/script/alpha_boxplot.R -h
# 完整参数,多样性指数可选richness chao1 ACE shannon simpson invsimpson
Rscript ${db}/script/alpha_boxplot.R --alpha_index richness \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
# 使用循环绘制6种常用指数
for i in `head -n1 result/alpha/vegan.txt|cut -f 2-`;do
Rscript ${db}/script/alpha_boxplot.R --alpha_index ${i} \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
done
mv alpha_boxplot_TukeyHSD.txt result/alpha/
# Alpha多样性柱状图+标准差
Rscript ${db}/script/alpha_barplot.R --alpha_index richness \
--input result/alpha/vegan.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 89 --height 59
### 1.2 稀释曲线
Rscript ${db}/script/alpha_rare_curve.R \
--input result/alpha/alpha_rare.txt --design result/metadata.txt \
--group Group --output result/alpha/ \
--width 120 --height 59
### 1.3 多样性维恩图
# 三组比较:-f输入文件,-a/b/c/d/g分组名,-w/u为宽高英寸,-p输出文件名后缀
bash ${db}/script/sp_vennDiagram.sh \
-f result/alpha/otu_group_exist.txt \
-a WT -b KO -c OE \
-w 3 -u 3 \
-p WT_KO_OE
# 四组比较,图和代码见输入文件目录,运行目录为当前项目根目录
bash ${db}/script/sp_vennDiagram.sh \
-f result/alpha/otu_group_exist.txt \
-a WT -b KO -c OE -d All \
-w 3 -u 3 \
-p WT_KO_OE_All
# EVenn在线绘制维恩图 https://www.ehbio.com/test/venn
## 2. Beta多样性
### 2.1 距离矩阵热图pheatmap
# 添加分组注释,如2,4列的基因型和地点
cut -f 1-2 result/metadata.txt > temp/group.txt
# 以bray_curtis为例,-f输入文件,-h是否聚类TRUE/FALSE,-u/v为宽高英寸
# -P添加行注释文件,-Q添加列注释
bash ${db}/script/sp_pheatmap.sh \
-f result/beta/bray_curtis.txt \
-H 'TRUE' -u 6.9 -v 5.6 \
-P temp/group.txt -Q temp/group.txt
### 2.2 主坐标分析PCoA
# 输入文件,选择分组,输出文件,图片尺寸mm,统计见beta_pcoa_stat.txt
Rscript ${db}/script/beta_pcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label FALSE --width 89 --height 59 \
--output result/beta/bray_curtis.pcoa.pdf
# 添加样本标签 --label TRUE
Rscript ${db}/script/beta_pcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label TRUE --width 89 --height 59 \
--output result/beta/bray_curtis.pcoa.label.pdf
mv beta_pcoa_stat.txt result/beta/
### 2.3 限制性主坐标分析CPCoA
Rscript ${db}/script/beta_cpcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --output result/beta/bray_curtis.cpcoa.pdf \
--width 89 --height 59
# 添加样本标签 --label TRUE
Rscript ${db}/script/beta_cpcoa.R \
--input result/beta/bray_curtis.txt --design result/metadata.txt \
--group Group --label TRUE --width 89 --height 59 \
--output result/beta/bray_curtis.cpcoa.label.pdf
## 3. 物种组成Taxonomy
### 3.1 堆叠柱状图Stackplot
# 以门(p)水平为例,结果包括output.sample/group.pdf两个文件
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_p.txt --design result/metadata.txt \
--group Group --color ggplot --legend 7 --width 89 --height 59 \
--output result/tax/sum_p.stackplot
# 修改颜色--color ggplot, manual1(22), Paired(12) or Set3(12)
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_p.txt --design result/metadata.txt \
--group Group --color Paired --legend 12 --width 181 --height 119 \
--output result/tax/sum_p.stackplotPaired
# 批量绘制输入包括p/c/o/f/g共5级
for i in p c o f g; do
Rscript ${db}/script/tax_stackplot.R \
--input result/tax/sum_${i}.txt --design result/metadata.txt \
--group Group --output result/tax/sum_${i}.stackplot \
--legend 8 --width 89 --height 59; done
### 3.2 弦/圈图circlize
# 以纲(class,c)为例,绘制前5组
i=c
Rscript ${db}/script/tax_circlize.R \
--input result/tax/sum_${i}.txt --design result/metadata.txt \
--group Group --legend 5
# 结果位于当前目录circlize.pdf(随机颜色),circlize_legend.pdf(指定颜色+图例)
# 移动并改名与分类级一致
mv circlize.pdf result/tax/sum_${i}.circlize.pdf
mv circlize_legend.pdf result/tax/sum_${i}.circlize_legend.pdf
### 3.3 树图treemap(参考)
# 多层级包含物种关系,输入特征表和物种注释,输出树图
# 指定包含特征数量和图片宽高,100个ASV耗时12s
Rscript ${db}/script/tax_maptree.R \
--input result/otutab.txt --taxonomy result/taxonomy.txt \
--output result/tax/tax_maptree.pdf \
--topN 100 --width 183 --height 118
# 24、差异比较
## 1. R语言差异分析
### 1.1 差异比较
# Error in file(file, ifelse(append, "a", "w")),输出目录不存在,创建目录即可
mkdir -p result/compare/
# 输入特征表、元数据;指定分组列名、比较组和丰度
# 选择方法 wilcox/t.test/edgeR、pvalue和fdr和输出目录
compare="KO-WT"
Rscript ${db}/script/compare.R \
--input result/otutab.txt --design result/metadata.txt \
--group Group --compare ${compare} --threshold 0.1 \
--method edgeR --pvalue 0.05 --fdr 0.2 \
--output result/compare/
### 1.2 火山图
# 输入compare.R的结果,输出火山图带数据标签,可指定图片大小
Rscript ${db}/script/compare_volcano.R \
--input result/compare/${compare}.txt \
--output result/compare/${compare}.volcano.pdf \
--width 89 --height 59
### 1.3 热图
# 输入compare.R的结果,筛选列数,指定元数据和分组、物种注释,图大小英寸和字号
bash ${db}/script/compare_heatmap.sh -i result/compare/${compare}.txt -l 7 \
-d result/metadata.txt -A Group \
-t result/taxonomy.txt \
-w 8 -h 5 -s 7 \
-o result/compare/${compare}
### 1.4 曼哈顿图
# i差异比较结果,t物种注释,p图例,w宽,v高,s字号,l图例最大值
# 图例显示不图,可增加高度v为119+即可,后期用AI拼图为KO-WT.heatmap.emf
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_p.txt \
-w 183 -v 59 -s 7 -l 10 \
-o result/compare/${compare}.manhattan.p.pdf
# 上图只有6个门,切换为纲c和-L Class展示细节
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_c.txt \
-w 183 -v 59 -s 7 -l 10 -L Class \
-o result/compare/${compare}.manhattan.c.pdf
# 显示完整图例,再用AI拼图
bash ${db}/script/compare_manhattan.sh -i result/compare/${compare}.txt \
-t result/taxonomy.txt \
-p result/tax/sum_c.txt \
-w 183 -v 149 -s 7 -l 10 -L Class \
-o result/compare/${compare}.manhattan.c.legend.pdf
### 1.5 单个特征的绘制
# 筛选显示差异ASV,按KO组丰度降序列,取ID展示前10
awk '$4<0.05' result/compare/KO-WT.txt | sort -k7,7nr | cut -f1 | head
# 差异OTU细节展示
Rscript ${db}/script/alpha_boxplot.R --alpha_index ASV_2 \
--input result/otutab.txt --design result/metadata.txt \
--transpose TRUE --scale TRUE \
--width 89 --height 59 \
--group Group --output result/compare/feature_
# ID不存在会报错: Error in data.frame(..., check.names = FALSE) : 参数值意味着不同的行数: 0, 18 Calls: alpha_boxplot -> cbind -> cbind -> data.frame
# 指定某列排序:按属丰度均值All降序
csvtk -t sort -k All:nr result/tax/sum_g.txt | head
# 差属细节展示
Rscript ${db}/script/alpha_boxplot.R --alpha_index Lysobacter \
--input result/tax/sum_g.txt --design result/metadata.txt \
--transpose TRUE \
--width 89 --height 59 \
--group Group --output result/compare/feature_
### 1.5 三元图
#参考示例见:result\compare\ternary\ternary.Rmd 文档
#备选教程[246.三元图的应用与绘图实战](https://mp.weixin.qq.com/s/3w3ncpwjQaMRtmIOtr2Jvw)
## 2. STAMP输入文件准备
### 2.1 生成输入文件
Rscript ${db}/script/format2stamp.R -h
mkdir -p result/stamp
Rscript ${db}/script/format2stamp.R --input result/otutab.txt \
--taxonomy result/taxonomy.txt --threshold 0.01 \
--output result/stamp/tax
# 可选Rmd文档见result/format2stamp.Rmd
### 2.2 绘制扩展柱状图和表
compare="KO-WT"
# 替换ASV(result/otutab.txt)为属(result/tax/sum_g.txt)
Rscript ${db}/script/compare_stamp.R \
--input result/stamp/tax_5Family.txt --metadata result/metadata.txt \
--group Group --compare ${compare} --threshold 0.1 \
--method "t.test" --pvalue 0.05 --fdr "none" \
--width 189 --height 159 \
--output result/stamp/${compare}
# 可选Rmd文档见result/CompareStamp.Rmd
## 3. LEfSe输入文件准备
### 3.1. 命令行生成文件
# 可选命令行生成输入文件
Rscript ${db}/script/format2lefse.R -h
mkdir -p result/lefse
# threshold控制丰度筛选以控制作图中的枝数量
Rscript ${db}/script/format2lefse.R --input result/otutab.txt \
--taxonomy result/taxonomy.txt --design result/metadata.txt \
--group Group --threshold 0.4 \
--output result/lefse/LEfSe
### 3.2 Rmd生成输入文件(可选)
#1. result目录中存在otutab.txt, metadata.txt, taxonomy.txt三个文件;
#2. Rstudio打开EasyAmplicon中format2lefse.Rmd,另存至result目录并Knit生成输入文件和可重复计算网页;
### 3.3 LEfSe分析
#方法1. 打开LEfSe.txt并在线提交 https://www.bic.ac.cn/BIC/#/analysis?page=b%27MzY%3D%27
#方法2. LEfSe本地分析(限Linux系统、选学),参考代码见附录
#方法3. LEfSe官网在线使用
# 25、QIIME 2分析流程
# 代码详见 qiime2/pipeline_qiime2.sh
# 31、功能预测
## 1. PICRUSt功能预测
### PICRUSt 1.0
# 方法1. 使用 http://www.ehbio.com/ImageGP 在线分析 gg/otutab.txt
# 方法2. Linux服务器用户可参考"附录2. PICRUSt功能预测"实现软件安装和分析
# 然后结果使用STAMP/R进行差异比较
# R语言绘图
# 输入文件格式调整
l=L2
sed '/# Const/d;s/OTU //' result/picrust/all_level.ko.${l}.txt > result/picrust/${l}.txt
num=`head -n1 result/picrust/${l}.txt|wc -w`
paste <(cut -f $num result/picrust/${l}.txt) <(cut -f 1-$[num-1] result/picrust/${l}.txt) \
> result/picrust/${l}.spf
cut -f 2- result/picrust/${l}.spf > result/picrust/${l}.mat.txt
awk 'BEGIN{FS=OFS="\t"} {print $2,$1}' result/picrust/${l}.spf | sed 's/;/\t/' | sed '1 s/ID/Pathway\tCategory/' \
> result/picrust/${l}.anno.txt
# 差异比较
compare="KO-WT"
Rscript ${db}/script/compare.R \
--input result/picrust/${l}.mat.txt --design result/metadata.txt \
--group Group --compare ${compare} --threshold 0 \
--method wilcox --pvalue 0.05 --fdr 0.2 \
--output result/picrust/
# 可对结果${compare}.txt筛选
# 绘制指定组(A/B)的柱状图,按高分类级着色和分面
Rscript ${db}/script/compare_hierarchy_facet.R \
--input result/picrust/${compare}.txt \
--data MeanA \
--annotation result/picrust/${l}.anno.txt \
--output result/picrust/${compare}.MeanA.bar.pdf
# 绘制两组显著差异柱状图,按高分类级分面
Rscript ${db}/script/compare_hierarchy_facet2.R \
--input result/picrust/${compare}.txt \
--pvalue 0.05 --fdr 0.1 \
--annotation result/picrust/${l}.anno.txt \
--output result/picrust/${compare}.bar.pdf
### PICRUSt 2.0
# 软件安装见附录6. PICRUSt环境导出和导入
# (可选)PICRUSt2(Linux/Windows下Linux子系统,要求>16GB内存)
# 安装参考附录5的方式直接下载安装包并解压即可使用
# Linux中加载conda环境
conda activate picrust2
# 进入工作目录,服务器要修改工作目录
wd=/mnt/d/amplicon/result/picrust2
mkdir -p ${wd} && cd ${wd}
# 运行流程,内存15.7GB,耗时12m
picrust2_pipeline.py -s ../otus.fa -i ../otutab.txt -o ./out -p 8
# 添加EC/KO/Pathway注释
cd out
add_descriptions.py -i pathways_out/path_abun_unstrat.tsv.gz -m METACYC \
-o METACYC.tsv
add_descriptions.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv.gz -m EC \
-o EC.tsv
add_descriptions.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv.gz -m KO \
-o KO.tsv
# KEGG按层级合并
db=/mnt/d/EasyMicrobiome/
python3 ${db}/script/summarizeAbundance.py \
-i KO.tsv \
-m ${db}/kegg/KO1-4.txt \
-c 2,3,4 -s ',+,+,' -n raw \
-o KEGG
# 统计各层级特征数量
wc -l KEGG*
# 可视化见picrust2文件夹中ggpicrust2.Rmd
## 2. 元素循环FAPROTAX
## 方法1. 在线分析,推荐使用 http://www.bic.ac.cn/ImageGP/index.php/Home/Index/FAPROTAX.html 在线分析
## 方法2. Linux下分析、如QIIME 2环境下
# 设置工作目录
wd=/mnt/d/amplicon/result/faprotax/
mkdir -p ${wd} && cd ${wd}
# 设置脚本目录
sd=/mnt/d/EasyMicrobiome/script/FAPROTAX_1.2.7
### 1. 软件安装
# 注:软件已经下载至 EasyMicrobiome/script目录,在qiime2环境下运行可满足依赖关系
#(可选)下载软件新版本,以1.2.7版为例, 2023/7/14更新数据库
#wget -c https://pages.uoregon.edu/slouca/LoucaLab/archive/FAPROTAX/SECTION_Download/MODULE_Downloads/CLASS_Latest%20release/UNIT_FAPROTAX_1.2.7/FAPROTAX_1.2.7.zip
#解压
#unzip FAPROTAX_1.2.7.zip
#新建一个python3环境并配置依赖关系,或进入qiime2 python3环境
conda activate qiime2-2023.7
# source /home/silico_biotech/miniconda3/envs/qiime2/bin/activate
#测试是否可运行,弹出帮助即正常工作
python $sd/collapse_table.py
### 2. 制作输入OTU表
#txt转换为biom json格式
biom convert -i ../otutab_rare.txt -o otutab_rare.biom --table-type="OTU table" --to-json
#添加物种注释
biom add-metadata -i otutab_rare.biom --observation-metadata-fp ../taxonomy2.txt \
-o otutab_rare_tax.biom --sc-separated taxonomy \
--observation-header OTUID,taxonomy
#指定输入文件、物种注释、输出文件、注释列名、属性列名
### 3. FAPROTAX功能预测
#python运行collapse_table.py脚本、输入带有物种注释OTU表tax.biom、
#-g指定数据库位置,物种注释列名,输出过程信息,强制覆盖结果,结果文件和细节
#下载faprotax.txt,配合实验设计可进行统计分析
#faprotax_report.txt查看每个类别中具体来源哪些OTUs
python ${sd}/collapse_table.py -i otutab_rare_tax.biom \
-g ${sd}/FAPROTAX.txt \
--collapse_by_metadata 'taxonomy' -v --force \
-o faprotax.txt -r faprotax_report.txt
### 4. 制作OTU对应功能注释有无矩阵
# 对ASV(OTU)注释行,及前一行标题进行筛选
grep 'ASV_' -B 1 faprotax_report.txt | grep -v -P '^--$' > faprotax_report.clean
# faprotax_report_sum.pl脚本将数据整理为表格,位于public/scrit中
perl ${sd}/../faprotax_report_sum.pl -i faprotax_report.clean -o faprotax_report
# 查看功能有无矩阵,-S不换行
less -S faprotax_report.mat
## 3. Bugbase细菌表型预测
### 1. Bugbase命令行分析
cd ${wd}/result
bugbase=${db}/script/BugBase
rm -rf bugbase/
# 脚本已经优化适合R4.0,biom包更新为biomformat
Rscript ${bugbase}/bin/run.bugbase.r -L ${bugbase} \
-i gg/otutab.txt -m metadata.txt -c Group -o bugbase/
### 2. 其它可用分析
# 使用 http://www.bic.ac.cn/ImageGP/index.php/Home/Index/BugBase.html
# 官网,https://bugbase.cs.umn.edu/ ,有报错,不推荐
# Bugbase细菌表型预测Linux,详见附录3. Bugbase细菌表型预测
# 32、MachineLearning机器学习
# RandomForest包使用的R代码见advanced/RandomForestClassification和RandomForestRegression
## Silme2随机森林/Adaboost使用代码见EasyMicrobiome/script/slime2目录中的slime2.py,详见附录4
# 使用实战(使用QIIME 2的Python3环境,以在Windows中为例)
conda activate qiime2-2023.7
cd /mnt/d/EasyMicrobiome/script/slime2
#使用adaboost计算10000次(16.7s),推荐千万次
./slime2.py otutab.txt design.txt --normalize --tag ab_e4 ab -n 10000
#使用RandomForest计算10000次(14.5s),推荐百万次,支持多线程
./slime2.py otutab.txt design.txt --normalize --tag rf_e4 rf -n 10000
# 33、Evolution进化树
cd ${wd}
mkdir -p result/tree
cd ${wd}/result/tree
## 1. 筛选高丰度/指定的特征
#方法1. 按丰度筛选特征,一般选0.001或0.005,且OTU数量在30-150个范围内
#统计特征表中ASV数量,如总计1609个
tail -n+2 ../otutab_rare.txt | wc -l
#按相对丰度0.2%筛选高丰度OTU
usearch -otutab_trim ../otutab_rare.txt \
-min_otu_freq 0.002 \
-output otutab.txt
#统计筛选OTU表特征数量,总计~81个
tail -n+2 otutab.txt | wc -l
#方法2. 按数量筛选
# #按丰度排序,默认由大到小
# usearch -otutab_sortotus ../otutab_rare.txt \
# -output otutab_sort.txt
# #提取高丰度中指定Top数量的OTU ID,如Top100,
# sed '1 s/#OTU ID/OTUID/' otutab_sort.txt \
# | head -n101 > otutab.txt
#修改特征ID列名
sed -i '1 s/#OTU ID/OTUID/' otutab.txt
#提取ID用于提取序列
cut -f 1 otutab.txt > otutab_high.id
# 筛选高丰度菌/指定差异菌对应OTU序列
usearch -fastx_getseqs ../otus.fa -labels otutab_high.id \
-fastaout otus.fa
head -n 2 otus.fa
## 筛选OTU对物种注释
awk 'NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}' ../taxonomy.txt \
otutab_high.id > otutab_high.tax
#获得OTU对应组均值,用于样本热图
#依赖之前otu_mean.R计算过按Group分组的均值
awk 'NR==FNR{a[$1]=$0} NR>FNR{print a[$1]}' ../otutab_mean.txt otutab_high.id \
| sed 's/#OTU ID/OTUID/' > otutab_high.mean
head -n3 otutab_high.mean
#合并物种注释和丰度为注释文件
cut -f 2- otutab_high.mean > temp
paste otutab_high.tax temp > annotation.txt
head -n 3 annotation.txt
## 2. 构建进化树
# 起始文件为 result/tree目录中 otus.fa(序列)、annotation.txt(物种和相对丰度)文件
# Muscle软件进行序列对齐,3s
muscle -in otus.fa -out otus_aligned.fas
### 方法1. 利用IQ-TREE快速构建ML进化树,2m
rm -rf iqtree
mkdir -p iqtree
iqtree -s otus_aligned.fas \
-bb 1000 -redo -alrt 1000 -nt AUTO \
-pre iqtree/otus
### 方法2. FastTree快速建树(Linux)
# 注意FastTree软件输入文件为fasta格式的文件,而不是通常用的Phylip格式。输出文件是Newick格式。
# 该方法适合于大数据,例如几百个OTUs的系统发育树!
# Ubuntu上安装fasttree可以使用`apt install fasttree`
# fasttree -gtr -nt otus_aligned.fas > otus.nwk
## 3. 进化树美化
# 访问http://itol.embl.de/,上传otus.nwk,再拖拽下方生成的注释方案于树上即美化
## 方案1. 外圈颜色、形状分类和丰度方案
# annotation.txt OTU对应物种注释和丰度,
# -a 找不到输入列将终止运行(默认不执行)-c 将整数列转换为factor或具有小数点的数字,-t 偏离提示标签时转换ID列,-w 颜色带,区域宽度等, -D输出目录,-i OTU列名,-l OTU显示名称如种/属/科名,
# cd ${wd}/result/tree
Rscript ${db}/script/table2itol.R -a -c double -D plan1 -i OTUID -l Genus -t %s -w 0.5 annotation.txt
# 生成注释文件中每列为单独一个文件
## 方案2. 生成丰度柱形图注释文件
Rscript ${db}/script/table2itol.R -a -d -c none -D plan2 -b Phylum -i OTUID -l Genus -t %s -w 0.5 annotation.txt
## 方案3. 生成热图注释文件
Rscript ${db}/script/table2itol.R -c keep -D plan3 -i OTUID -t %s otutab.txt
## 方案4. 将整数转化成因子生成注释文件
Rscript ${db}/script/table2itol.R -a -c factor -D plan4 -i OTUID -l Genus -t %s -w 0 annotation.txt
# 树iqtree/otus.contree在 http://itol.embl.de/ 上展示,拖拽不同Plan中的文件添加树注释
# 返回工作目录
cd ${wd}
## 4. 进化树可视化
https://www.bic.ac.cn/BIC/#/ 提供了更简易的可视化方式
# 附加视频
# 目录 Supp,网课有对应视频(可能编号不同,找关键字)
## S1. 网络分析R/CytoGephi
# 目录 Supp/S1NetWork
## S2. 溯源和马尔可夫链
# 目录 Supp/S2SourcetrackerFeastMarkov
## S11、网络分析ggClusterNet
# 代码:advanced/ggClusterNet/Practice.Rmd
## S12、Microeco包数据可视化
# 代码:advanced/microeco/Practice.Rmd
# 附录:Linux服务器下分析(选学)
#注:Windows下可能无法运行以下代码,推荐在Linux,或Windows下Linux子系统下conda安装相关程序
## 1. LEfSe分析
mkdir -p ~/amplicon/lefse
cd ~/amplicon/lefse
# format2lefse.Rmd代码制作或上传输入文件LEfSe.txt
# 安装lefse
# conda install lefse
#格式转换为lefse内部格式
lefse-format_input.py LEfSe.txt input.in -c 1 -o 1000000
#运行lefse
run_lefse.py input.in input.res
#绘制物种树注释差异
lefse-plot_cladogram.py input.res cladogram.pdf --format pdf
#绘制所有差异features柱状图
lefse-plot_res.py input.res res.pdf --format pdf
#绘制单个features柱状图(同STAMP中barplot)
head input.res #查看差异features列表
lefse-plot_features.py -f one --feature_name "Bacteria.Firmicutes.Bacilli.Bacillales.Planococcaceae.Paenisporosarcina" \
--format pdf input.in input.res Bacilli.pdf
#批量绘制所有差异features柱状图,慎用(几百张差异结果柱状图阅读也很困难)
mkdir -p features
lefse-plot_features.py -f diff --archive none --format pdf \
input.in input.res features/
## 2. PICRUSt功能预测
#推荐使用 http://www.bic.ac.cn/BIC/#/analysis?tool_type=tool&page=b%27Mzk%3D%27 在线分析
#有Linux服务器用户可参考以下代码搭建本地流程
n=picrust
conda create -n ${n} ${n} -c bioconda -y
wd=/mnt/d/amplicon
cd $wd/result/gg
# 启动环境
conda activate picrust
#上传gg/otutab.txt至当前目录
#转换为OTU表通用格式,方便下游分析和统计
biom convert -i otutab.txt \
-o otutab.biom \
--table-type="OTU table" --to-json
# 设置数据库目录,如 /mnt/d/db
db=~/db
#校正拷贝数,30s, 102M
normalize_by_copy_number.py -i otutab.biom \
-o otutab_norm.biom \
-c ${db}/picrust/16S_13_5_precalculated.tab.gz
#预测宏基因组KO表,3m,1.5G,biom方便下游归类,txt方便查看分析
predict_metagenomes.py -i otutab_norm.biom \
-o ko.biom \
-c ${db}/picrust/ko_13_5_precalculated.tab.gz
predict_metagenomes.py -f -i otutab_norm.biom \
-o ko.txt \
-c ${db}/picrust/ko_13_5_precalculated.tab.gz
#按功能级别分类汇总, -c输出KEGG_Pathways,分1-3级
sed -i '/# Constru/d;s/#OTU //' ko.txt
num=`head -n1 ko.txt|wc -w`
paste <(cut -f $num ko.txt) <(cut -f 1-$[num-1] ko.txt) > ko.spf
for i in 1 2 3;do
categorize_by_function.py -f -i ko.biom -c KEGG_Pathways -l ${i} -o pathway${i}.txt
sed -i '/# Const/d;s/#OTU //' pathway${i}.txt
paste <(cut -f $num pathway${i}.txt) <(cut -f 1-$[num-1] pathway${i}.txt) > pathway${i}.spf
done
wc -l *.spf
## 3. Bugbase细菌表型预测
### 1. 软件安装(己整合到EasyMicrobiome中,原代码需要更新才能在当前运行)
#有两种方法可选,推荐第一种,可选第二种,仅需运行一次
# #方法1. git下载,需要有git
# git clone https://github.com/knights-lab/BugBase
# #方法2. 下载并解压
# wget -c https://github.com/knights-lab/BugBase/archive/master.zip
# mv master.zip BugBase.zip
# unzip BugBase.zip
# mv BugBase-master/ BugBase
cd BugBase
#安装依赖包
export BUGBASE_PATH=`pwd`
export PATH=$PATH:`pwd`/bin
#安装了所有依赖包
run.bugbase.r -h
#测试数据
run.bugbase.r -i doc/data/HMP_s15.txt -m doc/data/HMP_map.txt -c HMPBODYSUBSITE -o output
### 2. 准备输入文件
cd ~/amplicon/result
#输入文件:基于greengene OTU表的biom格式(本地分析支持txt格式无需转换)和mapping file(metadata.txt首行添加#)
#上传实验设计+刚才生成的otutab_gg.txt
#生成在线分析使用的biom1.0格式
biom convert -i gg/otutab.txt -o otutab_gg.biom --table-type="OTU table" --to-json
sed '1 s/^/#/' metadata.txt > MappingFile.txt
#下载otutab_gg.biom 和 MappingFile.txt用于在线分析
### 3. 本地分析
export BUGBASE_PATH=`pwd`
export PATH=$PATH:`pwd`/bin
run.bugbase.r -i otutab_gg.txt -m MappingFile.txt -c Group -o phenotype/
## 4. Silme2随机森林/Adaboost
#下载安装
# cd ~/software/
# wget https://github.com/swo/slime2/archive/master.zip
# mv master.zip slime2.zip
# unzip slime2.zip
# mv slime2-master/ slime2
# cp slime2/slime2.py ~/bin/
# chmod +x ~/bin/slime2.py
#安装依赖包
# sudo pip3 install --upgrade pip
# sudo pip3 install pandas
# sudo pip3 install sklearn
## 5. PICRUSt2环境导出和导入
# 方法1. 下载安装包并解压
# 下载安装包,备用链接见百度云:https://pan.baidu.com/s/1Ikd_47HHODOqC3Rcx6eJ6Q?pwd=0315
wget -c ftp://download.nmdc.cn/tools/conda/picrust2.tar.gz
# 指定安装目录并解压
mkdir -p ~/miniconda3/envs/picrust2
tar -xvzf picrust2.tar.gz -C ~/miniconda3/envs/picrust2
# 激活环境并初始化
conda activate picrust2
conda unpack
# 方法2. 直接安装或打包安装环境
n=picrust2
conda create -n ${n} -c bioconda -c conda-forge ${n}=2.3.0_b
# 加载环境
conda activate ${n}
# 打包环境(可选)
conda pack -n ${n} -o ${n}.tar.gz
# 常见问题
## 1. 文件phred质量错误——Fastq质量值64转33
# 使用head查看fastq文件,phred64质量值多为小写字母,需要使用vsearch的--fastq_convert命令转换为通用的phred33格式。
cd /c/amplicon/FAQ/01Q64Q33
# 预览phred64格式,注意看第4行质量值多为小写字母
head -n4 test_64.fq
# 转换质量值64编码格式为33
vsearch --fastq_convert test_64.fq \
--fastq_ascii 64 --fastq_asciiout 33 \
--fastqout test.fq
# 查看转换后33编码格式,质量值多为大写字母
head -n4 test.fq
# 如果是Ion torrent测序结果,由于是非主流测序平台,需要公司转换帮助转换为标准的Phred33格式文件才可以使用。
## 2. 序列双端已经合并——单端序列添加样本名
# 扩增子分析要求序列名为样品名+序列编号,双端序列在合并同时可直接添加样本名。单端序列,或双端合并的序列需单独添加。这里使用vsearch的--fastq_convert命令中的--relabel参加添加样本名
cd /c/amplicon/FAQ/02relabel
# 查看文件序列名
head -n1 test.fq
# 序列按样本重命名
vsearch --fastq_convert test.fq \
--relabel WT1. \
--fastqout WT1.fq
# 查看重命名结果
head -n1 WT1.fq
## 3. 数据过大无法使用usearch聚类或去噪,替换vsearch
# 仅限usearch免费版受限时,可通过提高minuniquesize参数减少非冗余数据量。OTU/ASV过万下游分析等待时间过长,确保OTU/ASV数据小于5000,一般不会受限,而且也有利于下游开展快速分析。
# 备选vsearch聚类生成OTU,但无自动de novo去嵌合功能。输入2155条序列,聚类后输出661。
cd /c/amplicon/FAQ/03feature
# 重命名relabel、按相似id=97%聚类,不屏蔽qmask
# 记录输入sizein和输出频率sizeout
vsearch --cluster_size uniques.fa \
--relabel OTU_ --id 0.97 \
--qmask none --sizein --sizeout \
--centroids otus_raw.fa
# 再de novo去嵌合。55个嵌合,606个非嵌合。把OTU_1都去除了,没有Usearch内置去嵌合的方法合理。
# 自身比对去嵌合
vsearch --uchime_denovo otus_raw.fa \
--nonchimeras otus.fa
# 删除序列频率
sed -i 's/;.*//' otus.fa
## 4. 读长计数(Read counts)标准化为相对丰度
cd /c/amplicon/FAQ/04norm
# 求取各个OTU在样品中的丰度频率(标准化为总和1)
usearch -otutab_counts2freqs otutab.txt \
-output otutab_freq.txt
## 5. 运行R提示Permission denied
# 例如write.table保存表时,报错信息示例如下:意思是写入文件无权限,一般为目标文件正在被打开,请关闭相关文件后重试
Error in file(file, ifelse(append, "a", "w")) :
Calls: write.table -> file
: Warning message:
In file(file, ifelse(append, "a", "w")) :
'result/raw/otutab_nonBac.txt': Permission denied
## 6. 文件批量命名
# 如我们有文件A1和A2,编写一个样本名对应目标名的表格metadata.txt,检查样本名是否唯一,使用awk进行批量改名
cd /c/amplicon/FAQ/06rename
# (可选)快速生成文件列表,用于编辑metadata.txt,如A1.fq修改为WT1.fastq,以此类推,参考metadata.bak.txt
ls *.fq > metadata.txt
# 编辑列表,第二名为最终命名,确定名称唯一
# 转换行尾换行符
sed -i 's/\r//' metadata.txt
# 检查手动命名列2是否唯一
cut -f 2 metadata.txt|wc -l
cut -f 2 metadata.txt|sort|uniq|wc -l
# 如果两次结果一致,则命名非冗余
# 可选移动mv,复制cp,硬链ln,或软链ln -s
# 此处使用复制cp
awk '{system("cp "$1" "$2)}' metadata.txt
## 7. Rstudio中Terminal找不到Linux命令
# 需要把 C:\Program Files\Git\usr\bin 目录添加到系统环境变量
# 文件资源管理器——此电脑——属性——高级系统设置——环境变量——系统变量——Path——编辑——新建——填写“C:\Program Files\Git\usr\bin”——确定——确定——确定
# 注意win10系统是一个目录一行;win7中多个目录用分号分隔,注意向后添加目录
## 8. usearch -alpha_div_rare结果前两行出现“-”
#问题:抽样0时补“-”,且缺失制表符
#处理:替换“-”为"制作符\t+0"即可恢复
cd /c/amplicon/FAQ/08rare
sed "s/-/\t0.0/g" alpha_rare_wrong.txt\
> alpha_rare.txt
## 9. 物种注释otus.sintax方向全为“-”,需要序列取反向互补
#是原始序列方向错误,将filtered.fa序列需要取反向互补。再从头开始分析
cd /c/amplicon/FAQ/09revcom
vsearch --fastx_revcomp filtered_RC.fa \
--fastaout filtered.fa
## 10. windows换行符查看和删除
#Windows换行符为换行($)+^M,等于Linux换行+mac换行。分析数据中以linux格式为通用标准,因此windows中如excel编写并存为文本文件(制表符分隔)(*.txt)的表格,行尾有不可见的^M符号,导致分析出错。可使用cat -A命令查看此符号,可用sed删除。
cd /c/amplicon/FAQ/10^M
# 查看行尾是否有^M
cat -A metadata.txt
# 删除^M,并写入新文件
sed 's/\r//' metadata.txt > metadata.mod.txt
# 检查是否成功
cat -A metadata.mod.txt
# 直接原文件删除
sed -i 's/\r//' metadata.txt
## 11. UNITE数据库分析报错
#USEARCH使用UNITE下载的utax数据库,提示各种错误
cd /c/amplicon/FAQ/11unite
# 解压Unite的useach使用物种注释库
gunzip -c utax_reference_dataset_all_04.02.2020.fasta.gz > unite.fa
# 对ITS序列进行注释,默认阈值0.8
usearch --sintax otus.fa \
--db unite.fa \
--tabbedout otus.sintax --strand plus
--sintax_cutoff 0.6
#报错信息如下:
---Fatal error---
Missing x: in name >JN874928|SH1144646.08FU;tax=d:Metazoa,p:Cnidaria,c:Hydrozoa,o:Trachylina,f:,g:Craspedacusta,s:Craspedacusta_sowerbii_SH1144646.08FU;
“Unprintable ASCII character no 195 on or right before line 236492”
# 分析原因为分类级存在空缺。可用sed补全即可解决
# 分类级存在空缺,sed补全
sed -i 's/,;/,Unnamed;/;s/:,/:Unnamed,/g' unite.fa
# 再运行前面usearch --sintax命令
#注:vsearch有问题,推荐用usearch,结尾添加--strand plus才能成功运行
## 12. Windows的Linux子系统本地安装qiime2
# 详见 qiime2/pipeline_qiime2.sh
n=qiime2-2023.2
# 安装包下载链接
wget -c ftp://download.nmdc.cn/tools/conda/${n}.tar.gz
# 新环境安装
mkdir -p ~/miniconda3/envs/${n}
tar -xzf ${n}.tar.gz -C ~/miniconda3/envs/${n}
# 激活并初始化环境
conda activate ${n}
conda unpack
## 13. RDP 16-18注释结果比较
# 统计序列中门的数量,从60降为39
grep '>' ${db}/usearch/rdp_16s_v16_sp.fa|cut -f2 -d ';'|cut -f1-2 -d ','|sort|uniq|wc -l
grep '>' ${db}/usearch/rdp_16s_v18.fa|cut -f2 -d ';'|cut -f1-2 -d ','|sort|uniq|wc -l
# 统计序列中属的数量,从2517增长为3061
grep '>' ${db}/usearch/rdp_16s_v16_sp.fa|cut -f2 -d ';'|cut -f1-6 -d ','|sort|uniq|wc -l
grep '>' ${db}/usearch/rdp_16s_v18.fa|cut -f2 -d ';'|cut -f1-6 -d ','|sort|uniq|wc -l
cd /c/amplicon/FAQ/13rdp16_18
# 门由15个降为13个
tail -n+2 rdp16_sintax.txt|cut -f3|sort|uniq -c|wc -l
tail -n+2 rdp18_sintax.txt|cut -f3|sort|uniq -c|wc -l
# 属由176个降为144个
tail -n+2 rdp16_sintax.txt|cut -f7|sort|uniq -c|wc -l
tail -n+2 rdp18_sintax.txt|cut -f7|sort|uniq -c|wc -l
## 14. usearch生成OTU表小样本比vsearch更快
# usearch生成特征表,小样本(<30)快;但大样本受限且多线程效率低,83.2%, 4核17s
time usearch -otutab temp/filtered.fa \
-otus result/raw/otus.fa \
-threads 4 \
-otutabout result/raw/otutab.txt
# vsearch比对,更准更慢,但并行24-96线程更强
vsearch --usearch_global temp/filtered.fa --db ${db}/gg/97_otus.fa \
--otutabout result/gg/otutab.txt --id 0.97 --threads 12
# 比对率81.04%, 1核30m, 12核7m
# 版本更新记录
- 2021/4/3 EasyAmplicon 1.11:
- R包amplicon升级为 1.11.0,解决metadata两列报错的问题。
- 调整课程顺序,每天上午9点-12点2节,下午1点半-6点3节。
- 提供附加课程Supp目录。
- 2021/7/23 EasyAmplicon 1.12:
- R运行环境升级为4.1.0,配套有4.1.zip的全套包
- R包amplicon升级为 1.12.0,alpha_boxplot去掉Y轴的index
- alpha_boxplot.R增加标准化、转置等参数,可用于绘制任何特征箱线图
- beta_pcoa/cpcoa.R增加控制椭圆、标签显示等参数
- tax_stackplot.R增加多种配色方案
- picurst流程更新,并提供打包的conda下载
- picurst2新增KO合并为KEGG通路1-3级代码,并提供打包的conda下载
- 随机森林:提供分类级筛选、随机数筛选、可视化代码
- 2021/10/15 EasyAmplicon 1.13:
- R运行环境升级为4.1.1,配套有4.1.zip的最新全套包
- 元数据方差分解PERMANOVA:在Diversity-tutorial.Rmd中Beta多样性分析中新增adonis计算变量对群落的解析率和显著性分析
- 树图treemap升级后无颜色,改为代码供参考,并在Diversity_tutrial.Rmd中删除此部分
- alpha_boxplot输出无默认目录,可指定文件名头,添加无ID报错注释
- 2022/1/7 EasyAmplicon 1.14:
- R运行环境升级为4.1.2,配套有4.1.zip的最新全套包
- RStudio更新为2021.09.1
- 文涛重写amplicon包中tax_maptree函数,不依赖其他包,解决无法着色问题
- EasyMicrobiome中添加compare_stamp.R脚本,直接差异比较绘制STAMP扩展柱状图;代码详见result/CompareStamp.Rmd
- EasyMicrobiome中添加compare_hierarchy_facet.R和compare_hierarchy_facet2.R,展示KEGG的1,2级总览和差异
- 更新高级分析目录advanced:包括环境因子、马尔可无链、网络模块、网络比较、随机森林分类、随机森林回归、微生态等
- 2023/2/3 EasyAmplicon 1.18:
- R运行环境升级为4.2.2,配套有4.2.zip的最新全套包
- RStudio更新为2022.12.0
- amplicon、EasyAmplicon和EasyMicrobiome更新为1.18
- QIIME 2更新为v2023.2
- vsearch更新为v2.22.1
- 新增ggClusterNet课程-文涛
- 2023/10/13 EasyAmplicon 1.20:
- R运行环境升级为4.3.1,配套有4.3.zip的最新全套包
- RStudio更新为2023.12.0
- amplicon、EasyAmplicon和EasyMicrobiome更新为1.20
- QIIME 2更新为v2023.7,数据库更新为greengene2 2022.10
- 新增ggpicrust2分析picrust2结果可视化
- 更新FAPROTAX为1.2.7
每季度视频课程安排:http://www.ehbio.com/trainLongTerm/TrainLongTerm/amplicongenomeLearnGuide.html
使用此脚本,请引用下文:
If used this script, please cited:
Yong-Xin Liu, Lei Chen, Tengfei Ma, et al. 2023.
EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research.
iMeta 2: e83. https://doi.org/10.1002/imt2.83
Copyright 2016-2023 Yong-Xin Liu <liuyongxin@caas.cn>, Tao Wen <taowen@njau.edu.cn>, Tong Chen <chent@nrc.ac.cn>宏基因组学分析流程,绝对值得参考
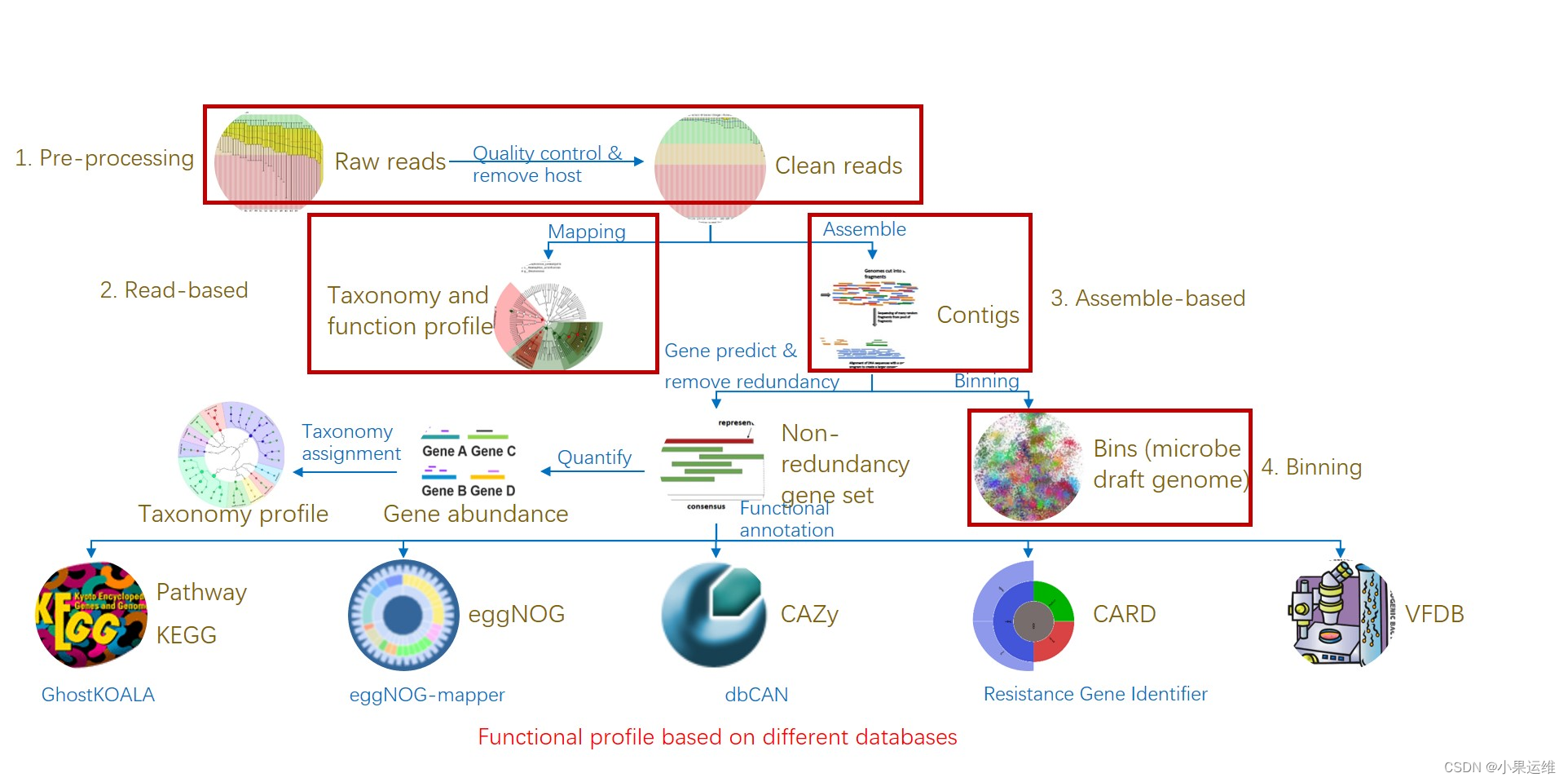
# 易宏基因组流程 EasyMetagenome Pipeline
# 版本Version: 1.20, 2023/11/23
# 操作系统Operation System: Linux Ubuntu 22.04+ / CentOS 7.7+
# 一、数据预处理 Data preprocessing
## 1.1 准备工作 Preparing
1. 首次使用请参照`0Install.sh`脚本,安装软件和数据库(大约1-3天,仅一次)
2. 易宏基因组(EasyMetagenome)流程`1Pipeline.sh`复制到项目文件夹,如本次为meta
3. 项目文件夹准备测序数据(seq/*.fq.gz)和样本元数据(result/metadata.txt)
**环境变量设置 Environment variable settings**
**分析前必须运行,设置数据库、软件和工作目录**
# Conda软件安装目录,`conda env list`查看,如/anaconda3
soft=~/miniconda3
# 数据库database(db)位置,如管理员/db,个人~/db
db=~/db
# 设置工作目录work directory(wd),如meta
wd=~/meta
# 创建并进入工作目录
mkdir -p $wd && cd $wd
# 创建3个常用子目录:序列,临时文件和结果
mkdir -p seq temp result
# 添加分析所需的软件、脚本至环境变量,添加至~/.bashrc中自动加载
PATH=$soft/bin:$soft/condabin:/usr/local/sbin:/usr/local/bin:/usr/sbin:/usr/bin:/sbin:/bin:$db/EasyMicrobiome/linux:$db/EasyMicrobiome/script
echo $PATH
**元数据和序列文件 Metadata and Sequence Files**
元数据
# 上传元数据metadata.txt至result目录,此处下载并重命名
wget http://www.imeta.science/github/EasyMetagenome/result/metadata.txt
mv metadata.txt result/metadata.txt
# 检查文件格式,^I为制表符,$为Linux换行,^M$为Windows回车,^M为Mac换行符
cat -A result/metadata.txt
# 根据样本文件生成元数据,可筛选子集,如EB开头
ls seq/EB*|grep '_1'|cut -f1 -d '_'|cut -f 2 -d '/'|sed'1 i SampleID'>result/metadataEB.txt
cp result/metadataEB.txt result/metadata.txt
# 元数据细节优化
# 转换Windows回车为Linux换行
sed -i 's/\r//' result/metadata.txt
# 去除数据中的一个多余空格
sed -i 's/Male /Male/' result/metadata.txt
cat -A result/metadata.txt
序列文件
# 用户使用filezilla上传测序文件至seq目录,本次从网络下载
# seq 目录下已经有测试文件,下载跳过
cd seq/
awk '{system("wget -c http://www.imeta.science/github/EasyMetagenome/seq/"$1"_1.fq.gz")}' <(tail -n+2 ../result/metadata.txt)
awk '{system("wget -c http://www.imeta.science/github/EasyMetagenome/seq/"$1"_2.fq.gz")}' <(tail -n+2 ../result/metadata.txt)
cd ..
# ls查看文件大小,-l 列出详细信息 (l: list),-sh 显示人类可读方式文件大小 (s: size; h: human readable)
ls -lsh seq/*.fq.gz
序列文件格式检查
zless/zcat查看可压缩文件,检查序列质量格式(质量值大写字母为标准Phred33格式,小写字母为Phred64,需参考附录:质量值转换);检查双端序列ID是否重名,如重名需要改名。参考**附录 —— 质控kneaddata,去宿主后双端不匹配;序列改名**。
# 设置某个样本名为变量i,以后再无需修改
i=C1
# zless查看压缩文件,空格翻页,q退出; head指定显示行数
zless seq/${i}_1.fq.gz | head -n4
**工作目录和文件结构总结**
# ├── pipeline.sh
# ├── result
# │ └── metadata.txt
# ├── seq
# │ ├── C1_1.fq.gz
# │ ├── ...
# │ └── N1_2.fq.gz
# └── temp
* 1pipeline.sh是分析流程代码;
* seq目录中有2个样本Illumina双端测序,4个序列文件;
* temp是临时文件夹,存储分析中间文件,结束可全部删除节约空间
* result是重要节点文件和整理化的分析结果图表,
* 实验设计metadata.txt也在此
## 1.2 Fastp质量控制 Quality Control
# 创建目录,记录软件版本和引文
mkdir -p temp/qc result/qc
fastp
# 单样本质控
i=C1
fastp -i seq/${i}_1.fq.gz -I seq/${i}_2.fq.gz \
-o temp/qc/${i}_1.fastq -O temp/qc/${i}_2.fastq
# 多样本并行
# -j 2: 表示同时处理2个样本
time tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \
"fastp -i seq/{1}_1.fq.gz -I seq/{1}_2.fq.gz \
-j temp/qc/{1}_fastp.json -h temp/qc/{1}_fastp.html \
-o temp/qc/{1}_1.fastq -O temp/qc/{1}_2.fastq \
> temp/qc/{1}.log 2>&1 "
# 质控后结果汇总
echo -e "SampleID\tRaw\tClean" > temp/fastp
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
echo -e -n "$i\t" >> temp/fastp
grep 'total reads' temp/qc/${i}.log|uniq|cut -f2 -d ':'|tr '\n' '\t' >> temp/fastp
echo "" >> temp/fastp
done
sed -i 's/ //g;s/\t$//' temp/fastp
# 按metadata排序
awk 'BEGIN{FS=OFS="\t"}NR==FNR{a[$1]=$0}NR>FNR{print a[$1]}' temp/fastp result/metadata.txt \
> result/qc/fastp.txt
cat result/qc/fastp.txt
## 1.3 KneadData去宿主 Host removal
kneaddata是流程主要依赖bowtie2比对宿主,然后筛选非宿主序列用于下游分析。
# 创建目录、启动环境、记录版本
mkdir -p temp/hr
conda activate kneaddata
kneaddata --version # 0.12.0
多样品并行去宿主,16p 4h
time tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \
"sed '1~4 s/ 1:/.1:/;1~4 s/$/\/1/' temp/qc/{}_1.fastq > /tmp/{}_1.fastq; \
sed '1~4 s/ 2:/.1:/;1~4 s/$/\/2/' temp/qc/{}_2.fastq > /tmp/{}_2.fastq; \
kneaddata -i1 /tmp/{1}_1.fastq -i2 /tmp/{1}_2.fastq \
-o temp/hr --output-prefix {1} \
--bypass-trim --bypass-trf --reorder \
--bowtie2-options '--very-sensitive --dovetail' \
-db ${db}/kneaddata/human/hg37dec_v0.1 \
--remove-intermediate-output -v -t 3; \
rm /tmp/{}_1.fastq /tmp/{}_2.fastq"
# * 匹配任意多个字符,? 匹配任意一个字符
ls -shtr temp/hr/*_paired_?.fastq
简化改名
# Ubuntu系统改名
rename 's/paired_//' temp/hr/*.fastq
# CentOS系统改名
rename 'paired_' '' temp/hr/*.fastq
大文件清理,高宿主含量样本可节约>90%空间
# 使用命令的绝对路径确保使用无参数的命令,管理员用alias自定义命令含参数,影响操作结果
/bin/rm -rf temp/hr/*contam* temp/hr/*unmatched* temp/hr/reformatted* temp/hr/_temp*
ls -l temp/hr/
质控结果汇总
kneaddata_read_count_table --input temp/hr \
--output temp/kneaddata.txt
# 筛选重点结果列
cut -f 1,2,5,6 temp/kneaddata.txt | sed 's/_1_kneaddata//' > result/qc/sum.txt
# 对齐方式查看表格
csvtk -t pretty result/qc/sum.txt
# 二、基于读长分析 Read-based (HUMAnN3+MetaPhlAn4+Kraken2)
## 2.1 准备HUMAnN输入文件
HUMAnN要求双端序列合并的文件作为输入,for循环根据实验设计样本名批量双端序列合并。注意星号(\*)和问号(?),分别代表多个和单个字符。当然大家更不能溜号,行分割的代码行末有一个\\
mkdir -p temp/concat
# 双端合并为单个文件
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
cat temp/hr/${i}_?.fastq \
> temp/concat/${i}.fq; done
# 查看样品数量和大小
ls -shl temp/concat/*.fq
# 数据太大,计算时间长,可用head对单端分析截取20M序列,即3G,行数为80M行,详见附录:HUMAnN2减少输入文件加速
## 2.2 HUMAnN计算物种和功能组成
* 物种组成调用MetaPhlAn4
* 输入文件:temp/concat/*.fq 每个样品质控后双端合并后的fastq序列
* 输出文件:temp/humann3/ 目录下
* C1_pathabundance.tsv
* C1_pathcoverage.tsv
* C1_genefamilies.tsv
* 整合后的输出:
* result/metaphlan4/taxonomy.tsv 物种丰度表
* result/metaphlan4/taxonomy.spf 物种丰度表(用于stamp分析)
* result/humann3/pathabundance_relab_unstratified.tsv 通路丰度表
* result/humann3/pathabundance_relab_stratified.tsv 通路物种组成丰度表
* stratified(每个菌对此功能通路组成的贡献)和unstratified(功能组成)
启动humann3环境,检查数据库配置
conda activate humann3
# 备选source加载指定环境
# source ~/miniconda3/envs/humann3/bin/activate
mkdir -p temp/humann3
humann --version # v3.7
humann_config
单样本1.25M PE150运行测试,8p,2.5M,1\~2h;0.2M, 34m;0.1M,30m;0.01M,25m;16p,18m
i=C1
# 3p,26m; 数据库使用ssd缩短到19m;16p,8m
time humann --input temp/concat/${i}.fq --output temp/humann3 --threads 3 --metaphlan-options '--bowtie2db /db/metaphlan4 --index mpa_vOct22_CHOCOPhlAnSGB_202212 --offline'
多样本并行计算,测试数据约30m,推荐16p,3小时/样本
# 如果服务器性能好,请设置--threads值为8/16/32
tail -n+2 result/metadata.txt | cut -f1 | rush -j 2 \
"humann --input temp/concat/{1}.fq \
--output temp/humann3/ --threads 3 --metaphlan-options '--bowtie2db /db/metaphlan4 --index mpa_vOct22_CHOCOPhlAnSGB_202212 --offline'"
# 移动重要文件至humann3目录
# $(cmd) 与 `cmd` 通常是等价的;`cmd`写法更简单,但要注意反引号是键盘左上角ESC下面的按键,$(cmd)更通用,适合嵌套使用
for i in $(tail -n+2 result/metadata.txt | cut -f1); do
mv temp/humann3/${i}_humann_temp/${i}_metaphlan_bugs_list.tsv temp/humann3/
done
# 删除临时文件,极占用空间
/bin/rm -rf temp/concat/* temp/humann3/*_humann_temp
(可选)单独运行MetaPhlAn4
mkdir -p temp/humann3
i=C1
# 仅物种注释极快4p, 2m, 1m读取数据库
time metaphlan --input_type fastq temp/qc/${i}_1.fastq \
temp/humann3/${i}.txt --bowtie2db /db/metaphlan4 --index mpa_vOct22_CHOCOPhlAnSGB_202212 --offline \
--nproc 4
## 2.3 物种组成表
**样品结果合并**
mkdir -p result/metaphlan4
# 合并、修正样本名、预览
merge_metaphlan_tables.py temp/humann3/*_metaphlan_bugs_list.tsv | \
sed 's/_metaphlan_bugs_list//g' | tail -n+2 | sed '1 s/clade_name/ID/' | sed '2i #metaphlan4'> result/metaphlan4/taxonomy.tsv
csvtk -t stat result/metaphlan4/taxonomy.tsv
head -n5 result/metaphlan4/taxonomy.tsv
**转换为stamp的spf格式**
# metaphlan4较2增加更多unclassified和重复结果,用sort和uniq去除
metaphlan_to_stamp.pl result/metaphlan4/taxonomy.tsv \
|sort -r | uniq > result/metaphlan4/taxonomy.spf
head result/metaphlan4/taxonomy.spf
# STAMP不支持unclassified,需要过滤掉再使用
grep -v 'unclassified' result/metaphlan4/taxonomy.spf > result/metaphlan4/taxonomy2.spf
# 下载metadata.txt和taxonomy2.spf使用stamp分析
## 2.4 功能组成分析
功能组成样本合并合并
mkdir -p result/humann3
humann_join_tables --input temp/humann3 \
--file_name pathabundance \
--output result/humann3/pathabundance.tsv
# 样本名调整:删除列名多余信息
sed -i 's/_Abundance//g' result/humann3/pathabundance.tsv
# 统计和预览
csvtk -t stat result/humann3/pathabundance.tsv
head -n5 result/humann3/pathabundance.tsv
标准化为相对丰度relab(1)或百万比cpm(1,000,000)
humann_renorm_table \
--input result/humann3/pathabundance.tsv \
--units relab \
--output result/humann3/pathabundance_relab.tsv
head -n5 result/humann3/pathabundance_relab.tsv
分层结果:功能和对应物种表(stratified)和功能组成表(unstratified)
humann_split_stratified_table \
--input result/humann3/pathabundance_relab.tsv \
--output result/humann3/
### 差异比较和柱状图
两样本无法组间比较,在pcl层面替换为HMP数据进行统计和可视化。
* 输入数据:通路丰度表格 result/humann3/pathabundance.tsv和实验设计 result/metadata.txt
* 中间数据:包含分组信息的通路丰度表格文件 result/humann3/pathabundance.pcl
* 输出结果:result/humann3/associate.txt
在通路丰度中添加分组
## 提取样品列表
head -n1 result/humann3/pathabundance.tsv | sed 's/# Pathway/SampleID/' | tr '\t' '\n' > temp/header
## 对应分组,本示例分组为第2列($2),根据实际情况修改
awk 'BEGIN{FS=OFS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' result/metadata.txt temp/header | tr '\n' '\t'|sed 's/\t$/\n/' > temp/group
# 合成样本、分组+数据
cat <(head -n1 result/humann3/pathabundance.tsv) temp/group <(tail -n+2 result/humann3/pathabundance.tsv) \
> result/humann3/pathabundance.pcl
head -n5 result/humann3/pathabundance.pcl
tail -n5 result/humann3/pathabundance.pcl
组间比较,样本量少无差异,结果为4列的文件:通路名字,通路在各个分组的丰度,差异P-value,校正后的Q-value。
演示数据2样本无法统计,此处替换为HMP的结果演示统计和绘图(上传hmp\_pathabund.pcl,替换pathabundance.pcl为hmp\_pathabund.pcl)。
wget -c http://www.imeta.science/github/EasyMetagenome/result/humann2/hmp_pathabund.pcl
/bin/cp -f hmp_pathabund.pcl result/humann3/
# 设置输入文件名
pcl=result/humann3/hmp_pathabund.pcl
# 统计表格行、列数量
csvtk -t stat ${pcl}
head -n3 ${pcl} | cut -f 1-5
# 按分组KW检验,注意第二列的分组列名
humann_associate --input ${pcl} \
--focal-metadatum Group --focal-type categorical \
--last-metadatum Group --fdr 0.05 \
--output result/humann3/associate.txt
wc -l result/humann3/associate.txt
head -n5 result/humann3/associate.txt
barplot展示通路的物种组成,如:腺苷核苷酸合成
# 指定差异通路,如 P163-PWY,--sort sum metadata 按丰度和分组排序
path=P163-PWY
humann_barplot \
--input ${pcl} --focal-feature ${path} \
--focal-metadata Group --last-metadata Group \
--output result/humann3/barplot_${path}.pdf --sort sum metadata
### KEGG注释
支持GO、PFAM、eggNOG、level4ec、KEGG的D级KO等注释,详见`humann_regroup_table -h`。
# 转换基因家族为KO(uniref90_ko),可选eggNOG(uniref90_eggnog)或酶(uniref90_level4ec)
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
humann_regroup_table \
-i temp/humann3/${i}_genefamilies.tsv \
-g uniref90_ko \
-o temp/humann3/${i}_ko.tsv
done
# 合并,并修正样本名
humann_join_tables \
--input temp/humann3/ \
--file_name ko \
--output result/humann3/ko.tsv
sed -i '1s/_Abundance-RPKs//g' result/humann3/ko.tsv
tail result/humann3/ko.tsv
# 与pathabundance类似,可进行标准化renorm、分层stratified、柱状图barplot等操作
# 分层结果:功能和对应物种表(stratified)和功能组成表(unstratified)
humann_split_stratified_table \
--input result/humann3/ko.tsv \
--output result/humann3/
wc -l result/humann3/ko*
# KO合并为高层次L2, L1通路代码KO to level 1/2/3
summarizeAbundance.py \
-i result/humann3/ko_unstratified.tsv \
-m ${db}/EasyMicrobiome/kegg/KO1-4.txt \
-c 2,3,4 -s ',+,+,' -n raw \
-o result/humann3/KEGG
wc -l result/humann3/KEGG*
## 2.5 GraPhlAn图
# metaphlan2 to graphlan
conda activate humann2
export2graphlan.py --skip_rows 1,2 -i result/metaphlan4/taxonomy.tsv \
--tree temp/merged_abundance.tree.txt \
--annotation temp/merged_abundance.annot.txt \
--most_abundant 1000 --abundance_threshold 20 --least_biomarkers 10 \
--annotations 3,4 --external_annotations 7
# 参数说明见PPT,或运行 export2graphlan.py --help
# graphlan annotation
graphlan_annotate.py --annot temp/merged_abundance.annot.txt \
temp/merged_abundance.tree.txt temp/merged_abundance.xml
# output PDF figure, annoat and legend
graphlan.py temp/merged_abundance.xml result/metaphlan4/graphlan.pdf \
--external_legends
# GraPhlAn Plot(测试中)
graphlan_plot.r --input result/metaphlan4/taxonomy.spf \
--design result/metadata.txt --number 100 \
--group all --type heatmap \
--output result/metaphlan4/heatmap
## 2.6 LEfSe差异分析物种
* 输入文件:物种丰度表result/metaphlan2/taxonomy.tsv
* 输入文件:样品分组信息 result/metadata.txt
* 中间文件:整合后用于LefSe分析的文件 result/metaphlan2/lefse.txt,这个文件可以提供给www\.ehbio.com/ImageGP 用于在线LefSE分析
* LefSe结果输出:result/metaphlan2/目录下lefse开头和feature开头的文件
前面演示数据仅有2个样本,无法进行差异比较。下面使用result12目录中由12个样本生成的结果表进行演示
# 设置结果目录,自己的数据使用result,演示用result12
result=result12
# 如果没有,请下载演示数据
wget -c http://www.imeta.science/db/EasyMetagenome/result12.zip
unzip result12.zip
准备输入文件,修改样本品为组名(可手动修改)
# 提取样本行替换为每个样本一行,修改ID为SampleID
head -n1 $result/metaphlan2/taxonomy.tsv|tr '\t' '\n'|sed '1 s/ID/SampleID/' >temp/sampleid
head -n3 temp/sampleid
# 提取SampleID对应的分组Group(假设为metadata.txt中第二列$2),替换换行\n为制表符\t,再把行末制表符\t替换回换行
awk 'BEGIN{OFS=FS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' $result/metadata.txt temp/sampleid|tr '\n' '\t'|sed 's/\t$/\n/' >groupid
cat groupid
# 合并分组和数据(替换表头)
cat groupid <(tail -n+2 $result/metaphlan2/taxonomy.tsv) > $result/metaphlan2/lefse.txt
head -n3 $result/metaphlan2/lefse.txt
方法1. 推荐在线 <https://www.bic.ac.cn/ImageGP/> 中LEfSe一键分析
方法2. LEfSe命令行分析
conda activate lefse
result=result12
# 格式转换为lefse内部格式
lefse-format_input.py $result/metaphlan2/lefse.txt \
temp/input.in -c 1 -o 1000000
# 运行lefse(样本必须有重复和分组)
run_lefse.py temp/input.in temp/input.res
# 绘制物种树注释差异
lefse-plot_cladogram.py temp/input.res \
$result/metaphlan2/lefse_cladogram.pdf --format pdf
# 绘制所有差异features柱状图
lefse-plot_res.py temp/input.res \
$result/metaphlan2/lefse_res.pdf --format pdf
# 绘制单个features柱状图
# 查看显著差异features,按丰度排序
grep -v '-' temp/input.res | sort -k3,3n
# 手动选择指定feature绘图,如Firmicutes
lefse-plot_features.py -f one --format pdf \
--feature_name "k__Bacteria.p__Firmicutes" \
temp/input.in temp/input.res \
$result/metaphlan2/lefse_Firmicutes.pdf
# 批量绘制所有差异features柱状图
lefse-plot_features.py -f diff \
--archive none --format pdf \
temp/input.in temp/input.res \
$result/metaphlan2/lefse_
## 2.7 Kraken2+Bracken物种注释和丰度估计
Kraken2可以快速完成读长(read)层面的物种注释和定量,还可以进行重叠群(contig)、基因(gene)、宏基因组组装基因组(MAG/bin)层面的序列物种注释。
# 启动kraken2工作环境
conda activate kraken2
# 记录软件版本
kraken2 --version # 2.1.2
mkdir -p temp/kraken2
### Kraken2物种注释
输入:temp/qc/{1}_?.fastq 质控后的数据,{1}代表样本名;
参考数据库:-db ${db}/kraken2/pluspfp16g/
输出结果:每个样本单独输出,temp/kraken2/中的{1}_report和{1}_output
整合物种丰度表输出结果:result/kraken2/taxonomy_count.txt
(可选) 单样本注释,5m,50G大数据库较5G库注释比例提高10~20%。以C1为例,在2023/3/14版中,8g: 31.75%; 16g: 52.35%; 150g: 71.98%;同为16g,2023/10/9版本为63.88%
# 根据电脑内存由小到大选择下面3个数据库
# pluspf16g/pluspfp(55G)/plusppfp(120G)
i=C1
time kraken2 --db ${db}/kraken2/pluspf16g/ \
--paired temp/qc/${i}_?.fastq \
--threads 2 --use-names --report-zero-counts \
--report temp/kraken2/${i}.report \
--output temp/kraken2/${i}.output
多样本并行生成report,1样本8线程逐个运行,内存大但速度快,不建议用多任务并行
for i in `tail -n+2 result/metadata.txt | cut -f1`;do
kraken2 --db ${db}/kraken2/pluspf16g \
--paired temp/qc/${i}_?.fastq \
--threads 2 --use-names --report-zero-counts \
--report temp/kraken2/${i}.report \
--output temp/kraken2/${i}.output; done
使用krakentools转换report为mpa格式
for i in `tail -n+2 result/metadata.txt | cut -f1`;do
kreport2mpa.py -r temp/kraken2/${i}.report \
--display-header -o temp/kraken2/${i}.mpa; done
合并样本为表格
mkdir -p result/kraken2
# 输出结果行数相同,但不一定顺序一致,要重新排序
tail -n+2 result/metadata.txt | cut -f1 | rush -j 1 \
'tail -n+2 temp/kraken2/{1}.mpa | LC_ALL=C sort | cut -f 2 | sed "1 s/^/{1}\n/" > temp/kraken2/{1}_count '
# 提取第一样本品行名为表行名
header=`tail -n 1 result/metadata.txt | cut -f 1`
echo $header
tail -n+2 temp/kraken2/${header}.mpa | LC_ALL=C sort | cut -f 1 | \
sed "1 s/^/Taxonomy\n/" > temp/kraken2/0header_count
head -n3 temp/kraken2/0header_count
# paste合并样本为表格
ls temp/kraken2/*count
paste temp/kraken2/*count > result/kraken2/tax_count.mpa
# 检查表格及统计
csvtk -t stat result/kraken2/tax_count.mpa
head -n 5 result/kraken2/tax_count.mpa
### Bracken丰度估计
参数简介:
* -d为数据库,-i为输入kraken2报告文件
* r是读长,此处为100,通常为150,o输出重新估计的值
* l为分类级,可选域D、门P、纲C、目O、科F、属G、种S级别丰度估计
* t是阈值,默认为0,越大越可靠,但可用数据越少
循环重新估计每个样品的丰度,请修改tax分别重新计算P和S各1次
# 设置估算的分类级别D,P,C,O,F,G,S,常用门P和种S
# 测序6G起样本-t 10过滤低丰度物种
tax=S
mkdir -p temp/bracken
for i in `tail -n+2 result/metadata.txt | cut -f1`;do
# i=C1
bracken -d ${db}/kraken2/pluspf16g/ \
-i temp/kraken2/${i}.report \
-r 100 -l ${tax} -t 0 \
-o temp/bracken/${i}.brk \
-w temp/bracken/${i}.report; done
# bracken结果合并成表: 需按表头排序,提取第6列reads count,并添加样本名
tail -n+2 result/metadata.txt | cut -f1 | rush -j 1 \
'tail -n+2 temp/bracken/{1}.brk | LC_ALL=C sort | cut -f6 | sed "1 s/^/{1}\n/" \
> temp/bracken/{1}.count'
# 提取第一样本品行名为表行名
h=`tail -n1 result/metadata.txt|cut -f1`
tail -n+2 temp/bracken/${h}.brk | sort | cut -f1 | \
sed "1 s/^/Taxonomy\n/" > temp/bracken/0header.count
# 检查文件数,为n+1
ls temp/bracken/*count | wc
# paste合并样本为表格,并删除非零行
paste temp/bracken/*count > result/kraken2/bracken.${tax}.txt
# 统计行列,默认去除表头
csvtk -t stat result/kraken2/bracken.${tax}.txt
# 按频率过滤,-r可标准化,-e过滤(microbiome_helper)
Rscript ${db}/EasyMicrobiome/script/filter_feature_table.R \
-i result/kraken2/bracken.${tax}.txt \
-p 0.01 \
-o result/kraken2/bracken.${tax}.0.01
csvtk -t stat result/kraken2/bracken.${tax}.0.01
个性化结果筛选
# 门水平去除脊索动物(人)
grep 'Chordata' result/kraken2/bracken.P.0.01
grep -v 'Chordata' result/kraken2/bracken.P.0.01 > result/kraken2/bracken.P.0.01-H
# 按物种名手动去除宿主污染,以人为例(需按种水平计算相关结果)
# 种水平去除人类P:Chordata,S:Homo sapiens
grep 'Homo sapiens' result/kraken2/bracken.S.0.01
grep -v 'Homo sapiens' result/kraken2/bracken.S.0.01 \
> result/kraken2/bracken.S.0.01-H
分析后清理每条序列的注释大文件
/bin/rm -rf temp/kraken2/*.output
### 多样性和可视化
alpha多样性计算:Berger Parker’s (BP), Simpson’s (Si), inverse Simpson’s (ISi), Shannon’s (Sh) # Fisher’s (F)依赖scipy.optimize包,默认未安装
mkdir -p result/kraken2
echo -e "SampleID\tBerger Parker\tSimpson\tinverse Simpson\tShannon" > result/kraken2/alpha.txt
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
echo -e -n "$i\t" >> result/kraken2/alpha.txt
for a in BP Si ISi Sh;do
alpha_diversity.py -f temp/bracken/${i}.brk -a $a | cut -f 2 -d ':' | tr '\n' '\t' >> result/kraken2/alpha.txt
done
echo "" >> result/kraken2/alpha.txt
done
cat result/kraken2/alpha.txt
beta多样性计算
beta_diversity.py -i temp/bracken/*.brk --type bracken \
> result/kraken2/beta.txt
cat result/kraken2/beta.txt
Krona图
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
kreport2krona.py -r temp/bracken/${i}.report -o temp/bracken/${i}.krona --no-intermediate-ranks
ktImportText temp/bracken/${i}.krona -o result/kraken2/krona.${i}.html
done
Pavian桑基图:https://fbreitwieser.shinyapps.io/pavian/ 在线可视化:,左侧菜单,Upload sample set (temp/bracken/*.report),支持多样本同时上传;Sample查看结果,Configure Sankey配置图样式,Save Network下载图网页
多样性分析/物种组成,详见3StatPlot.sh,Kraken2结果筛选序列见附录
# 三、组装分析流程 Assemble-based
## 组装
# 启动工作环境
conda activate megahit
### MEGAHIT组装Assembly
# 删除旧文件夹,否则megahit无法运行
# /bin/rm -rf temp/megahit
# 组装,10~30m,TB级数据需几天至几周
megahit -t 3 \
-1 `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/hr\//;s/$/_1.fastq/'|tr '\n' ','|sed 's/,$//'` \
-2 `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/hr\//;s/$/_2.fastq/'|tr '\n' ','|sed 's/,$//'` \
-o temp/megahit
# 统计大小通常300M~5G,如果contigs太多,可以按长度筛选,降低数据量,提高基因完整度,详见附录megahit
seqkit stat temp/megahit/final.contigs.fa
# 预览重叠群最前6行,前60列字符
head -n6 temp/megahit/final.contigs.fa | cut -c1-60
# 备份重要结果
mkdir -p result/megahit/
ln -f temp/megahit/final.contigs.fa result/megahit/
# 删除临时文件
/bin/rm -rf temp/megahit/intermediate_contigs
### (可选)metaSPAdes精细组装
# 精细但使用内存和时间更多,15~65m
/usr/bin/time -v -o metaspades.py.log metaspades.py -t 3 -m 100 \
`tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_1.fastq/'|sed 's/^/-1 /'| tr '\n' ' '` \
`tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_2.fastq/'|sed 's/^/-2 /'| tr '\n' ' '` \
-o temp/metaspades
# 查看软件时间User time和内存Maximum resident set size
cat metaspades.py.log
# 2.3M,contigs体积更大
ls -sh temp/metaspades/contigs.fasta
seqkit stat temp/metaspades/contigs.fasta
# 备份重要结果
mkdir -p result/metaspades/
ln -f temp/metaspades/contigs.fasta result/metaspades/
# 删除临时文件
/bin/rm -rf temp/metaspades
注:metaSPAdes支持二、三代混合组装,见附录,此外还有OPERA-MS组装二、三代方案
---
### QUAST评估
# QUAST评估,生成report文本tsv/txt、网页html、PDF等格式报告
quast.py result/megahit/final.contigs.fa \
-o result/megahit/quast -t 2
# (可选) megahit和metaspades比较
quast.py --label "megahit,metapasdes" \
result/megahit/final.contigs.fa \
result/metaspades/contigs.fasta \
-o result/quast
# (可选)metaquast评估,更全面,但需下载相关数据库,受网速影响可能时间很长(经常失败)
# metaquast based on silva, and top 50 species genome, 18min
time metaquast.py result/megahit/final.contigs.fa \
-o result/megahit/metaquast
## 3.2 基因预测、去冗余和定量Gene prediction, cluster & quantitfy
### metaProdigal基因预测Gene prediction
# 输入文件:组装的序列 result/megahit/final.contigs.fa
# 输出文件:prodigal预测的基因序列 temp/prodigal/gene.fa
# 基因大,可参考附录prodigal拆分基因文件,并行计算
mkdir -p temp/prodigal
# prodigal的meta模式预测基因,>和2>&1记录分析过程至gene.log
prodigal -i result/megahit/final.contigs.fa \
-d temp/prodigal/gene.fa \
-o temp/prodigal/gene.gff \
-p meta -f gff > temp/prodigal/gene.log 2>&1
# 查看日志是否运行完成,有无错误
tail temp/prodigal/gene.log
# 统计基因数量
seqkit stat temp/prodigal/gene.fa
# 统计完整基因数量,数据量大可只用完整基因部分
grep -c 'partial=00' temp/prodigal/gene.fa
# 提取完整基因(完整片段获得的基因全为完整,如成环的细菌基因组)
grep 'partial=00' temp/prodigal/gene.fa | cut -f1 -d ' '| sed 's/>//' > temp/prodigal/full_length.id
seqkit grep -f temp/prodigal/full_length.id temp/prodigal/gene.fa > temp/prodigal/full_length.fa
seqkit stat temp/prodigal/full_length.fa
### cd-hit基因聚类/去冗余cluster & redundancy
# 输入文件:prodigal预测的基因序列 temp/prodigal/gene.fa
# 输出文件:去冗余后的基因和蛋白序列:result/NR/nucleotide.fa, result/NR/protein.fa
mkdir -p result/NR
# aS覆盖度,c相似度,G局部比对,g最优解,T多线程,M内存0不限制
# 2万基因2m,2千万需要2000h,多线程可加速
cd-hit-est -i temp/prodigal/gene.fa \
-o result/NR/nucleotide.fa \
-aS 0.9 -c 0.95 -G 0 -g 0 -T 0 -M 0
# 统计非冗余基因数量,单次拼接结果数量下降不大,多批拼接冗余度高
grep -c '>' result/NR/nucleotide.fa
# 翻译核酸为对应蛋白序列, --trim去除结尾的*
seqkit translate --trim result/NR/nucleotide.fa \
> result/NR/protein.fa
# 两批数据去冗余使用cd-hit-est-2d加速,见附录
### salmon基因定量quantitfy
# 输入文件:去冗余后的基因序列:result/NR/nucleotide.fa
# 输出文件:Salmon定量:result/salmon/gene.count, gene.TPM
mkdir -p temp/salmon
salmon -v # 1.8.0
# 建索引, -t序列, -i 索引,10s
salmon index -t result/NR/nucleotide.fa \
-p 3 -i temp/salmon/index
# 定量,l文库类型自动选择,p线程,--meta宏基因组模式, 2个任务并行2个样
tail -n+2 result/metadata.txt | cut -f1 | rush -j 2 \
"salmon quant -i temp/salmon/index -l A -p 3 --meta \
-1 temp/qc/{1}_1.fastq -2 temp/qc/{1}_2.fastq \
-o temp/salmon/{1}.quant"
# 合并
mkdir -p result/salmon
salmon quantmerge --quants temp/salmon/*.quant \
-o result/salmon/gene.TPM
salmon quantmerge --quants temp/salmon/*.quant \
--column NumReads -o result/salmon/gene.count
sed -i '1 s/.quant//g' result/salmon/gene.*
# 预览结果表格
head -n3 result/salmon/gene.*
## 3.3 功能基因注释Functional gene annotation
# 输入数据:上一步预测的蛋白序列 result/NR/protein.fa
# 中间结果:temp/eggnog/protein.emapper.seed_orthologs
# temp/eggnog/output.emapper.annotations
# temp/eggnog/output
# COG定量表:result/eggnog/cogtab.count
# result/eggnog/cogtab.count.spf (用于STAMP)
# KO定量表:result/eggnog/kotab.count
# result/eggnog/kotab.count.spf (用于STAMP)
# CAZy碳水化合物注释和定量:result/dbcan3/cazytab.count
# result/dbcan3/cazytab.count.spf (用于STAMP)
# 抗生素抗性:result/resfam/resfam.count
# result/resfam/resfam.count.spf (用于STAMP)
# 这部分可以拓展到其它数据库
### eggNOG基因注释gene annotation(COG/KEGG/CAZy)
软件主页:https://github.com/eggnogdb/eggnog-mapper
# 运行并记录软件版本
conda activate eggnog
emapper.py --version
# emapper-2.1.7 / Expected eggNOG DB version: 5.0.2
# Diamond version found: diamond version 2.0.15
# 运行emapper,18m,默认diamond 1e-3
mkdir -p temp/eggnog
time emapper.py --data_dir ${db}/eggnog \
-i result/NR/protein.fa --cpu 3 -m diamond --override \
-o temp/eggnog/output
# 格式化结果并显示表头
grep -v '^##' temp/eggnog/output.emapper.annotations | sed '1 s/^#//' \
> temp/eggnog/output
csvtk -t headers -v temp/eggnog/output
# 生成COG/KO/CAZy丰度汇总表
mkdir -p result/eggnog
# 显示帮助
summarizeAbundance.py -h
# 汇总,7列COG_category按字母分隔,12列KEGG_ko和19列CAZy按逗号分隔,原始值累加
summarizeAbundance.py \
-i result/salmon/gene.TPM \
-m temp/eggnog/output --dropkeycolumn \
-c '7,12,19' -s '*+,+,' -n raw \
-o result/eggnog/eggnog
sed -i 's#^ko:##' result/eggnog/eggnog.KEGG_ko.raw.txt
sed -i '/^-/d' result/eggnog/eggnog*
head -n3 result/eggnog/eggnog*
# eggnog.CAZy.raw.txt eggnog.COG_category.raw.txt eggnog.KEGG_ko.raw.txt
# 添加注释生成STAMP的spf格式
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \
${db}/EasyMicrobiome/kegg/KO_description.txt \
result/eggnog/eggnog.KEGG_ko.raw.txt | \
sed 's/^\t/Unannotated\t/' \
> result/eggnog/eggnog.KEGG_ko.TPM.spf
head -n 5 result/eggnog/eggnog.KEGG_ko.TPM.spf
# KO to level 1/2/3
summarizeAbundance.py \
-i result/eggnog/eggnog.KEGG_ko.raw.txt \
-m ${db}/EasyMicrobiome/kegg/KO1-4.txt \
-c 2,3,4 -s ',+,+,' -n raw --dropkeycolumn \
-o result/eggnog/KEGG
head -n3 result/eggnog/KEGG*
# CAZy
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \
${db}/EasyMicrobiome/dbcan2/CAZy_description.txt result/eggnog/eggnog.CAZy.raw.txt | \
sed 's/^\t/Unannotated\t/' > result/eggnog/eggnog.CAZy.TPM.spf
head -n 3 result/eggnog/eggnog.CAZy.TPM.spf
# COG
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2"\t"$3} NR>FNR{print a[$1],$0}' \
${db}/EasyMicrobiome/eggnog/COG.anno result/eggnog/eggnog.COG_category.raw.txt > \
result/eggnog/eggnog.COG_category.TPM.spf
head -n 3 result/eggnog/eggnog.COG_category.TPM.spf
### CAZy碳水化合物酶
# 比对CAZy数据库, 用时2~18m
mkdir -p temp/dbcan3 result/dbcan3
# --sensitive慢10倍,dbcan3e值为1e-102,此处以1e-3演示
time diamond blastp \
--db ${db}/dbcan3/CAZyDB \
--query result/NR/protein.fa \
--threads 2 -e 1e-3 --outfmt 6 --max-target-seqs 1 --quiet \
--out temp/dbcan3/gene_diamond.f6
wc -l temp/dbcan3/gene_diamond.f6
# 提取基因与dbcan分类对应表
format_dbcan2list.pl \
-i temp/dbcan3/gene_diamond.f6 \
-o temp/dbcan3/gene.list
# 按对应表累计丰度,依赖
summarizeAbundance.py \
-i result/salmon/gene.TPM \
-m temp/dbcan3/gene.list \
-c 2 -s ',' -n raw --dropkeycolumn \
-o result/dbcan3/TPM
# 添加注释生成STAMP的spf格式,结合metadata.txt进行差异比较
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \
${db}/EasyMicrobiome/dbcan2/CAZy_description.txt result/dbcan3/TPM.CAZy.raw.txt | \
sed 's/^\t/Unannotated\t/' \
> result/dbcan3/TPM.CAZy.raw.spf
head result/dbcan3/TPM.CAZy.raw.spf
# 检查未注释数量,有则需要检查原因
grep 'Unannotated' result/dbcan3/TPM.CAZy.raw.spf|wc -l
### CARD耐药基因
CARD在线分析平台:https://card.mcmaster.ca/
本地软件使用教程: https://github.com/arpcard/rgi
参考文献:http://doi.org/10.1093/nar/gkz935
结果说明:protein.json,在线可视化;protein.txt,注释基因列表
mkdir -p result/card
# 启动rgi环境和记录版本
conda activate rgi6
rgi main -v # 6.0.3
# 简化蛋白ID
cut -f 1 -d ' ' result/NR/protein.fa > temp/protein.fa
# 这个错误忽略即可,不是报错,没有任何影响 grep: 写错误: 断开的管道
grep '>' result/NR/protein.fa | head -n 3
grep '>' temp/protein.fa | head -n 3
# 蛋白层面注释ARG
rgi main -i temp/protein.fa -t protein \
-n 9 -a DIAMOND --include_loose --clean \
-o result/card/protein
head -n3 result/card/protein.txt
# 基因层面注释ARG
cut -f 1 -d ' ' result/NR/nucleotide.fa > temp/nucleotide.fa
grep '>' temp/nucleotide.fa | head -n3
rgi main -i temp/nucleotide.fa -t contig \
-n 9 -a DIAMOND --include_loose --clean \
-o result/card/nucleotide
head -n3 result/card/nucleotide.txt
# 重叠群层面注释ARG
cut -f 1 -d ' ' result/megahit/final.contigs.fa > temp/contigs.fa
grep '>' temp/contigs.fa | head -n3
rgi main -i temp/contigs.fa -t contig \
-n 9 -a DIAMOND --include_loose --clean \
-o result/card/contigs
head result/card/contigs.txt
## 3.4 基因物种注释
# Generate report in default taxid output
conda activate kraken2
kraken2 --db ${db}/kraken2/pluspf16g \
result/NR/nucleotide.fa \
--threads 3 \
--report temp/NRgene.report \
--output temp/NRgene.output
# Genes & taxid list
grep '^C' temp/NRgene.output | cut -f 2,3 | sed '1 i Name\ttaxid' \
> temp/NRgene.taxid
# Add taxonomy
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $1,a[$2]}' \
$db/EasyMicrobiome/kraken2/taxonomy.txt \
temp/NRgene.taxid > result/NR/nucleotide.tax
conda activate eggnog
summarizeAbundance.py \
-i result/salmon/gene.TPM \
-m result/NR/nucleotide.tax --dropkeycolumn \
-c '2,3,4,5,6,7,8,9' -s ',+,+,+,+,+,+,+,' -n raw \
-o result/NR/tax
wc -l result/NR/tax*|sort -n
# 四、分箱挖掘单菌基因组Binning
## 4.1 MetaWRAP混合样本分箱 Samples binning
主页:https://github.com/bxlab/metaWRAP
教程: https://github.com/bxlab/metaWRAP/blob/master/Usage_tutorial.md
挖掘单菌基因组,需要研究对象复杂度越低、测序深度越大,结果质量越好。要求单样本6GB+,复杂样本如土壤推荐数据量30GB+,至少3个样本
演示数据12个样仅140MB,无法获得单菌基因组,这里使用官方测序数据演示讲解
软件和数据库布置需1-3天,演示数据分析过程超10h,30G样也需1-30天,由服务器性能决定。
# 设置并进入工作目录
wd=~/meta/binning
mkdir -p ${wd} && cd ${wd}
# 初始化项目
mkdir -p temp/hr seq result
# 启动metawrap环境
conda activate metawrap
### 数据和环境变量 Data and enviroment
这里基于质控clean数据和拼接好的重叠群contigs,基于上游结果继续分析。由于上游测试数据过小,分箱无结果。 本次采用软件推荐的7G数据,我们进入一个新文件夹开展分析。
输入输出文件介绍:
# 输入:质控后序列,文件名格式为*_1.fastq和*_2.fastq,temp/qc 目录下,如C1_1.fastq、C1_2.fastq
# 组装的重叠群文件:result/megahit/final.contigs.fa
# 输出:
# Binning结果:temp/binning
# 提纯后的Bin统计结果:temp/bin_refinement/metawrap_50_10_bins.stats
# Bin定量结果文件和图:binning/temp/bin_quant/bin_abundance_table.tab 和 bin_abundance_heatmap.png
# Bin物种注释:binning/temp/bin_classify/bin_taxonomy.tab
# Prokka基因预测:binning/temp/bin_annotate/prokka_out/bin.*.ffn 核酸序列
# Bin可视化图表:binning/temp/bloblogy/final.contigs.binned.blobplot (数据表) 和 blobplot_figures (可视化图)
准备输入文件:原始数据+组装结果
# 质控后数据位于temp/qc中,此处需下载并解压
# 方法1. 直接拷贝
/bin/cp -rf /db/metawrap/*.fastq ~/meta/binning/temp/hr/
# 方法2. 在线下载
cd temp/hr
for i in `seq 7 9`;do
wget -c ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR01134${i}/ERR01134${i}_1.fastq.gz
wget -c ftp.sra.ebi.ac.uk/vol1/fastq/ERR011/ERR01134${i}/ERR01134${i}_2.fastq.gz
done
gunzip -k *.gz
# 批量修改扩展名fq为fastq
# rename .fq .fastq *.fq
# megahit拼接结果
cd ${wd}
mkdir -p temp/megahit
cd temp/megahit
# 可从EasyMetagenome目录复制,或链接下载
wget -c http://www.imeta.science/db/metawrap/final.contigs.fa.gz
gunzip -k *.gz
cd ${wd}
### 分箱Binning
# 加载运行环境
cd ${wd}
conda activate metawrap
metawrap -v # 1.3.2
# 输入文件为contig和clean reads
# 调用maxbin2, metabat2,8p线程2h,24p耗时1h;-concoct 3h
nohup metawrap binning -o temp/binning \
-t 3 -a temp/megahit/final.contigs.fa \
--metabat2 --maxbin2 \
temp/hr/*.fastq
# --concoct > /dev/null 2>&1 增加3~10倍计算量,添加/dev/null清除海量Warning信息
### 分箱提纯Bin refinement
# 8线程2h, 24p 1.3h;2方法16p 20m
metawrap bin_refinement \
-o temp/bin_refinement \
-A temp/binning/metabat2_bins/ \
-B temp/binning/maxbin2_bins/ \
-c 50 -x 10 -t 8
# -C temp/binning/concoct_bins/ \
# 统计高质量Bin的数量,2方法6个,3方法9个
tail -n+2 temp/bin_refinement/metawrap_50_10_bins.stats|wc -l
# 分析比较图见 temp/bin_refinement/figures/
所有分箱至同一目录All bins in one directory
mkdir -p temp/drep_in
# 混合组装分箱链接和重命名
ln -s `pwd`/temp/bin_refinement/metawrap_50_10_bins/bin.* temp/drep_in/
ls -l temp/drep_in/
# 改名CentOS
rename 'bin.' 'Mx_All_' temp/drep_in/bin.*
# 改名Ubuntu
rename s/bin./Mx_All_/ temp/drep_in/bin.*
ls temp/drep_in/Mx*
## (可选Opt)单样本分箱Single sample binning
多样本受硬件、计算时间限制无法完成时,需要单样本组装、分箱。多样本信息丰度,分箱结果更多,更容易降低污染。详见:- [Nature Methods | 单样本与多样本宏基因组分箱的比较揭示了广泛存在的隐藏性污染](https://mp.weixin.qq.com/s/i5C-rCVhZyjRK_Dsk36vBQ)
**设置全局线程、并行任务数和筛选分箱的条件**
# p:threads线程数,job任务数,complete完整度x:contaminate污染率
conda activate metawrap
p=16
j=3
c=50
x=10
(可选)并行需要样本列表,请提前编写metadata.txt保存于result中
# 快速读取文件生成样本ID列表再继续编写
ls temp/hr/ | grep _1 | cut -f 1 -d '_' | sort -u | sed '1 i SampleID' > result/metadata.txt
# 预览
cat result/metadata.txt
**组装Assemble**
单样本并行组装;支持中断继续运行
tail -n+2 result/metadata.txt|cut -f1|rush -j ${j} \
"metawrap assembly -m 200 -t ${p} --megahit \
-1 temp/hr/{}_1.fastq -2 temp/hr/{}_2.fastq \
-o temp/megahit/{}"
**分箱binning**
单样本并行分箱,192p, 15m (concoct使用超多线程);16p 2d/sample, >/dev/null 16p 12h/sample
time tail -n+2 result/metadata.txt|cut -f1|rush -j ${j} \
"metawrap binning \
-o temp/binning/{} -t ${p} \
-a temp/megahit/{}/final_assembly.fasta \
--metabat2 --maxbin2 --concoct \
temp/hr/{}_*.fastq > /dev/null 2>&1"
**分箱提纯bin refinement**
time tail -n+2 result/metadata.txt|cut -f1|rush -j ${j} \
"metawrap bin_refinement \
-o temp/bin_refinement/{} -t ${p} \
-A temp/binning/{}/metabat2_bins/ \
-B temp/binning/{}/maxbin2_bins/ \
-C temp/binning/{}/concoct_bins/ \
-c ${c} -x ${x} "
# 分别为1,2,2个
tail -n+2 result/metadata.txt|cut -f1|rush -j 1 \
"tail -n+2 temp/bin_refinement/{}/metawrap_50_10_bins.stats|wc -l "
单样品分箱链接和重命名
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
ln -s `pwd`/temp/bin_refinement/${i}/metawrap_50_10_bins/bin.* temp/drep_in/
# CentOS
rename 'bin.' "Sg_${i}_" temp/drep_in/bin.*
# Ubuntu
rename "s/bin./Sg_${i}_/" temp/drep_in/bin.*
done
# 删除空白中无效链接
/bin/rm -f temp/drep_in/*\*
# 统计混合和单样本来源数据,10个混,5个单;不同系统结果略有差异
ls temp/drep_in/|cut -f 1 -d '_'|uniq -c
# 统计混合批次/单样本来源
ls temp/drep_in/|cut -f 2 -d '_'|cut -f 1 -d '.' |uniq -c
## (可选Opt)分组分箱 Subgroup binning
样本>30或数据量>300G在1TB内存胖结点上完成混合组装和分箱可能内存不足、且时间>1周甚至1月,需要对研究相近条件、地点进行分小组,且每组编写一个metadata??.txt。
conda activate metawrap
# 小组ID: A1/A2/A3
g=A1
**组装Assemble**:<30个或<300G样本,~12h
metawrap assembly -m 600 -t 32 --megahit \
-1 `tail -n+2 result/metadata${g}.txt|cut -f1|sed 's/^/temp\/hr\//;s/$/_1.fastq/'|tr '\n' ','|sed 's/,$//'` \
-2 `tail -n+2 result/metadata${g}.txt|cut -f1|sed 's/^/temp\/hr\//;s/$/_2.fastq/'|tr '\n' ','|sed 's/,$//'` \
-o temp/megahit_${g}
**分箱Binning**,~18h
# 链接文件到临时位置
mkdir -p temp/${g}/
for i in `tail -n+2 result/metadata${g}.txt|cut -f1`;do
ln -s `pwd`/temp/hr/${i}*.fastq temp/${g}/
done
# 按组分箱
metawrap binning -o temp/binning_${g} \
-t 32 -a temp/megahit_${g}/final_assembly.fasta \
--metabat2 --maxbin2 \
temp/${g}/*.fastq
**分箱提纯Bin refinement**
metawrap bin_refinement \
-o temp/bin_refinement_${g} \
-A temp/binning_${g}/metabat2_bins/ \
-B temp/binning_${g}/maxbin2_bins/ \
-c 50 -x 10 -t 32
# 统计高质量Bin的数量
wc -l temp/bin_refinement_${g}/metawrap_50_10_bins.stats
**改名汇总 Rename & merge**
mkdir -p temp/drep_in
# 混合组装分箱链接和重命名
ln -s `pwd`/temp/bin_refinement_${g}/metawrap_50_10_bins/bin.* temp/drep_in/
# 改名
rename "s/bin./Gp_${g}_/" temp/drep_in/bin.* # Ubuntu
# rename 'bin.' "Gp_${g}_" temp/drep_in/bin.* # CentOS
# 统计
mkdir -p result/bin
echo -n $g >> result/bin/groupNo.txt
ls temp/drep_in/Gp_${g}_*|wc>> result/bin/groupNo.txt
cat result/bin/groupNo.txt
## 4.2 dRep去冗余种/株基因组集
# 进入虚拟环境drep和工作目录
conda activate drep
cd ${wd}
按种水平去冗余:6~40min,15个为10个,8个来自混拼,2个来自单拼
mkdir -p temp/drep95
# /bin/rm -rf temp/drep95/data/checkM
time dRep dereplicate temp/drep95/ \
-g temp/drep_in/*.fa \
-sa 0.95 -nc 0.30 -comp 50 -con 10 -p 5
# 报错日志在temp/drep95/log/cmd_logs中查看,加-d显示更多
ls temp/drep95/dereplicated_genomes/|cut -f 1 -d '_'|sort|uniq -c
主要结果temp/drep95中:
* 非冗余基因组集:temp/drep95/dereplicated_genomes/*.fa
* 聚类信息表:temp/drep95/data_tables/Cdb.csv
* 聚类和质量图:temp/drep95/figures/*clustering*
(可选)按株水平99%去冗余,20-30min,本处也为10个
mkdir -p temp/drep99
time dRep dereplicate temp/drep99/ \
-g temp/drep_in/*.fa \
-sa 0.99 -nc 0.30 -comp 50 -con 10 -p 5
ls -l temp/drep99/dereplicated_genomes/ | grep '.fa' | wc -l
## 4.3 CoverM基因组定量
# 启动环境
conda activate coverm
mkdir -p temp/coverm
# (可选)单样本测试, 3min
i=ERR011347
time coverm genome --coupled temp/hr/${i}_1.fastq temp/hr/${i}_2.fastq \
--genome-fasta-directory temp/drep95/dereplicated_genomes/ -x fa \
-o temp/coverm/${i}.txt
cat temp/coverm/${i}.txt
# 并行计算, 4min
tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \
"coverm genome --coupled temp/hr/{}_1.fastq temp/hr/{}_2.fastq \
--genome-fasta-directory temp/drep95/dereplicated_genomes/ -x fa \
-o temp/coverm/{}.txt"
# 结果合并
mkdir -p result/coverm
conda activate humann3
sed -i 's/_1.fastq Relative Abundance (%)//' temp/coverm/*.txt
humann_join_tables --input temp/coverm \
--file_name txt \
--output result/coverm/abundance.tsv
# 按组求均值,需要metadata中有3列且每个组有多个样本
Rscript ${db}/EasyMicrobiome/script/otu_mean.R --input result/coverm/abundance.tsv \
--metadata result/metadata.txt \
--group Group --thre 0 \
--scale TRUE --zoom 100 --all TRUE --type mean \
--output result/coverm/group_mean.txt
# https://www.bic.ac.cn/ImageGP/ 直接选择热图可视化
## 4.4 GTDB-tk物种注释和进化树
启动软件所在虚拟环境
conda activate gtdbtk2.3
export GTDBTK_DATA_PATH="${db}/gtdb"
gtdbtk -v # 2.3.2
代表性细菌基因组物种注释
mkdir -p temp/gtdb_classify
# 10个基因组,24p,100min 152G内存; 6p, 22基因组,1h
gtdbtk classify_wf \
--genome_dir temp/drep95/dereplicated_genomes \
--out_dir temp/gtdb_classify \
--extension fa --skip_ani_screen \
--prefix tax \
--cpus 6
# less -S按行查看,按q退出
less -S temp/gtdb_classify/tax.bac120.summary.tsv
less -S temp/gtdb_classify/tax.ar53.summary.tsv
代表种注释:以上面鉴定的10个种为例,注意扩展名要与输入文件一致,可使用压缩格式gz。主要结果文件描述:此9个细菌基因组在tax.bac120.summary.tsv。古菌在tax.ar53开头的文件中。
(可选)所有MAG物种注释
mkdir -p temp/gtdb_all
# 10000个基因组,32p,100min
time gtdbtk classify_wf \
--genome_dir temp/drep_in/ \
--out_dir temp/gtdb_all \
--extension fa --skip_ani_screen \
--prefix tax \
--cpus 6
less -S temp/gtdb_all/tax.bac120.summary.tsv
less -S temp/gtdb_all/tax.ar53.summary.tsv
多序列对齐结果建树
# 以9个细菌基因组的120个单拷贝基因建树,1s
mkdir -p temp/gtdb_infer
gtdbtk infer --msa_file temp/gtdb_classify/align/tax.bac120.user_msa.fasta.gz \
--out_dir temp/gtdb_infer --prefix tax --cpus 3
树文件`tax.unrooted.tree`可使用iTOL在线美化,也可使用GraphLan本地美化。
制作树注释文件:以gtdb-tk物种注释(tax.bac120.summary.tsv)和drep基因组评估(Widb.csv)信息为注释信息
mkdir -p result/itol
# 制作分类学表
tail -n+2 temp/gtdb_classify/tax.bac120.summary.tsv|cut -f 1-2|sed 's/;/\t/g'|sed '1 s/^/ID\tDomain\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' \
> result/itol/tax.txt
head result/itol/tax.txt
# 基因组评估信息
sed 's/,/\t/g;s/.fa//' temp/drep95/data_tables/Widb.csv|cut -f 1-7,11|sed '1 s/genome/ID/' \
> result/itol/genome.txt
head result/itol/genome.txt
# 整合注释文件
awk 'BEGIN{OFS=FS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $0,a[$1]}' result/itol/genome.txt result/itol/tax.txt|cut -f 1-8,10- > result/itol/annotation.txt
head result/itol/annotation.txt
# 添加各样本相对丰度(各组替换均值)
awk 'BEGIN{OFS=FS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $0,a[$1]}' <(sed '1 s/Genome/ID/' result/coverm/abundance.tsv) result/itol/annotation.txt|cut -f 1-15,17- > result/itol/annotation2.txt
head result/itol/annotation2.txt
### CheckM2重新评估
conda activate checkm2
mkdir -p temp/checkm2 result/checkm2
# 10 genomes, 2m
time checkm2 predict --input temp/drep95/dereplicated_genomes/* --output-directory temp/checkm2 --threads 8
ln temp/checkm2/quality_report.tsv result/checkm2/
# 查看结果
less result/checkm2//quality_report.tsv
# 5 (可选)单菌基因组
## 5.1 Fastp质量控制
# 每个样本~30s,173个j2共
mkdir -p temp/qc/
time tail -n+2 result/metadata.txt | cut -f1 | rush -j 2 \
"time fastp -i seq/{1}_1.fq.gz -I seq/{1}_2.fq.gz \
-j temp/qc/{1}_fastp.json -h temp/qc/{1}_fastp.html \
-o temp/qc/{1}_1.fastq -O temp/qc/{1}_2.fastq \
> temp/qc/{1}.log 2>&1"
## 5.2 metaspades组装
conda activate megahit
spades.py -v # v3.15.4,>3.14.0才支持--isolate模式
mkdir -p temp/spades result/spades
# 127 genoms, 1m17s
time tail -n+2 result/metadata.txt|cut -f1|rush -j 3 \
"spades.py --pe1-1 temp/qc/{1}_1.fastq \
--pe1-2 temp/qc/{1}_2.fastq \
-t 16 --isolate --cov-cutoff auto \
-o temp/spades/{1}"
# 筛选>1k的序列并汇总、统计
time tail -n+2 result/metadata.txt|cut -f1|rush -j 3 \
"seqkit seq -m 1000 temp/spades/{1}/contigs.fasta \
> temp/spades/{1}.fa"
seqkit stat temp/spades/*.fa | sed 's/temp\/spades\///;s/.fa//' > result/spades/stat1k.txt
## 5.3 checkm质量评估
checkm评估质量
conda activate drep
checkm # CheckM v1.1.2
mkdir -p temp/checkm result/checkm
# 127 genoms, 1m17s
time checkm lineage_wf -t 8 -x fa temp/spades/ temp/checkm
# format checkm jason to tab
checkmJason2tsv.R -i temp/checkm/storage/bin_stats_ext.tsv \
-o temp/checkm/bin_stats.txt
csvtk -t pretty temp/checkm/bin_stats.txt | less
(可选)checkm2评估(测试中...)
conda activate checkm2
mkdir -p temp/checkm2
time checkm2 predict --threads 8 --input temp/spades/ --output-directory temp/checkm2
筛选污染和高质量基因组 >5% contamination and high quailty
awk '$5<90 || $10>5' temp/checkm/bin_stats.txt | csvtk -t cut -f 1,5,10,4,9,2 > temp/checkm/contamination5.txt
tail -n+2 temp/checkm/contamination5.txt|wc -l
# 筛选高质量用于下游分析 <5% high-quality for down-stream analysis
awk '$5>=90 && $10<=5' temp/checkm/bin_stats.txt | csvtk -t cut -f 1,5,10,4,9,2 | sed '1 i ID\tCompleteness\tContamination\tGC\tN50\tsize' > result/checkm/Comp90Cont5.txt
tail -n+2 result/checkm/Comp90Cont5.txt|wc -l
# 链接高质量基因组至新目录,单菌完整度通常>99%
mkdir -p temp/drep_in/
for n in `tail -n+2 result/checkm/Comp90Cont5.txt|cut -f 1`;do
ln temp/spades/${n}.fa temp/drep_in/
done
## 5.4 混菌metawarp分箱
分箱和提纯binning & refinement
conda activate metawrap
mkdir -p temp/binning temp/bin
time tail -n+2 temp/checkm/contamination5.txt|cut -f1|rush -j 3 \
"metawrap binning \
-o temp/binning/{} -t 8 \
-a temp/spades/{}/contigs.fasta \
--metabat2 --maxbin2 \
temp/qc/{}_*.fastq"
time tail -n+2 temp/checkm/contamination5.txt|cut -f1|rush -j 15 \
"metawrap bin_refinement \
-o temp/bin/{} -t 8 \
-A temp/binning/{}/metabat2_bins/ \
-B temp/binning/{}/maxbin2_bins/ \
-c 50 -x 10"
分箱结果汇总
echo -n -e "" > temp/bin/metawrap.stat
for m in `tail -n+2 temp/checkm/contamination5.txt|cut -f1`;do
echo ${m} >> temp/bin/metawrap.stat
cut -f1-4,6-7 temp/bin/${m}/metawrap_50_10_bins.stats >> temp/bin/metawrap.stat
done
# 分箱后的按b1,b2,b3重命名共培养,单菌也可能减少污染
for m in `tail -n+2 temp/checkm/contamination5.txt|cut -f1`;do
c=1
for n in `tail -n+2 temp/bin/$m/metawrap_50_10_bins.stats|cut -f 1`;do
cp temp/bin/$m/metawrap_50_10_bins/${n}.fa temp/drep_in/${m}b${c}.fa
((c++))
done
done
分箱前后统计比较
# 如107个测序分箱为352个基因组,共418个基因组
tail -n+2 temp/checkm/contamination5.txt|wc -l
ls temp/drep_in/*b?.fa | wc -l
ls temp/drep_in/*.fa | wc -l
# 重建新ID列表,A代表所有,B代表Bin分箱过的单菌
ls temp/drep_in/*.fa|cut -f 3 -d '/'|sed 's/.fa//'|sed '1 i ID'|less -S>result/metadataA.txt
ls temp/drep_in/*b?.fa|cut -f 3 -d '/'|sed 's/.fa//'|sed '1 i ID'|less -S>result/metadataB.txt
可视化混菌中覆盖度分布,以第一污染菌为例
i=`tail -n+2 temp/checkm/contamination5.txt|head -n1|cut -f1`
# grep '>' temp/spades/${i}.fa|cut -f 2,4,6 -d '_'|sed 's/^/C/;s/_/\t/g'|sed '1i Contig\tLength\tCoverage'>temp/spades/${i}.len
grep '>' temp/drep_in/${i}*|cut -f 3 -d '/'|sed 's/.fa:>NODE//'|cut -f 1,2,4,6 -d '_'|sed 's/_/\t/g'|sed '1i Genome\tContig\tLength\tvalue' > temp/drep_in/${i}.cov
sp_lines.sh -f temp/drep_in/${i}.cov -m TRUE -A TRUE -a Contig -H Genome
## 5.5 drep基因组去冗余
mkdir -p temp/drep95/
conda activate drep
# 相似度sa 0.99995 去重复, 0.99 株水平, 0.95 种水平
dRep dereplicate \
-g temp/drep_in/*.fa \
-sa 0.95 -nc 0.3 -p 16 -comp 50 -con 10 \
temp/drep95
# 统计总和使用基因组数据,确保没有异常丢弃stat total and used genomes, keep no abnormal discard
ls temp/drep_in/*.fa|wc -l
grep 'passed checkM' temp/drep95/log/logger.log|sed 's/[ ][ ]*/ /g'|cut -f 4 -d ' '
# 去冗余后数量,418变为49种
ls temp/drep95/dereplicated_genomes/|wc -l
# 唯一和重复的基因组unique and duplicate genome
csvtk cut -f 11 temp/drep95/data_tables/Widb.csv | sort | uniq -c
# 整理种列表
echo "SampleID" > result/metadataS.txt
ls temp/drep95/dereplicated_genomes/|sed 's/\.fa//' >> result/metadataS.txt
## 5.6 gtdb物种注释
conda activate gtdbtk2.3
# 所有基因组注释,400g, 1h, 1T
mkdir -p temp/gtdb_all result/gtdb_all
memusg -t gtdbtk classify_wf \
--genome_dir temp/drep_in/ \
--out_dir temp/gtdb_all/ \
--extension fa --skip_ani_screen \
--prefix tax \
--cpus 16
# 基因组信息genomeInfo.csv
sed 's/,/\t/g;s/.fa//' temp/drep95/data_tables/genomeInfo.csv |sed '1 s/genome/ID/' > result/gtdb_all/genome.txt
# 95%聚类种基因组注释,40g, 1h, 500G
mkdir -p temp/gtdb_95 result/gtdb_95
# Taxonomy classify
memusg -t gtdbtk classify_wf \
--genome_dir temp/drep95/dereplicated_genomes/ \
--out_dir temp/gtdb_95 \
--extension fa --skip_ani_screen \
--prefix tax \
--cpus 8
# Phylogenetic tree infer
memusg -t gtdbtk infer \
--msa_file temp/gtdb_95/align/tax.bac120.user_msa.fasta.gz \
--out_dir temp/gtdb_95 \
--cpus 8 --prefix g >> temp/gtdb_95/infer.log 2>&1
ln `pwd`/temp/gtdb_95/infer/intermediate_results/g.unrooted.tree result/gtdb_95/
# 细菌format to standard 7 levels taxonomy
tail -n+2 temp/gtdb_95/classify/tax.bac120.summary.tsv|cut -f 1-2|sed 's/;/\t/g'|sed '1 s/^/ID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' > result/gtdb_95/tax.bac.txt
# 古菌(可选)
tail -n+2 temp/gtdb_95/classify/tax.ar122.summary.tsv|cut -f 1-2|sed 's/;/\t/g'|sed '1 s/^/ID\tKingdom\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' > result/gtdb_95/tax.ar.txt
cat result/gtdb_95/tax.bac.txt <(tail -n+2 result/gtdb_95/tax.ar.txt) > result/gtdb_95/tax.txt
# Widb.csv 非冗余基因组信息
sed 's/,/\t/g;s/.fa//' temp/drep95/data_tables/Widb.csv|cut -f 1-7,11|sed '1 s/genome/ID/' > result/gtdb_95/genome.txt
# 整合物种注释和基因组信息 Integrated taxonomy and genomic info
awk 'BEGIN{OFS=FS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $0,a[$1]}' result/gtdb_95/genome.txt result/gtdb_95/tax.txt|cut -f 1-8,10- > result/gtdb_95/annotation.txt
# csvtk -t headers -v result/gtdb_95/annotation.txt
# 制作itol files
cd result/gtdb_95
table2itol.R -D plan1 -a -c double -i ID -l Genus -t %s -w 0.5 annotation.txt
table2itol.R -D plan2 -a -d -c none -b Phylum -i ID -l Genus -t %s -w 0.5 annotation.txt
table2itol.R -D plan3 -c keep -i ID -t %s annotation.txt
table2itol.R -D plan4 -a -c factor -i ID -l Genus -t %s -w 0 annotation.txt
# Stat each level
echo -e 'Taxonomy\tKnown\tNew' > tax.stat
for i in `seq 2 8`;do
head -n1 tax.txt|cut -f ${i}|tr '\n' '\t' >> tax.stat
tail -n+2 tax.txt|cut -f ${i}|grep -v '__$'|sort|uniq -c|wc -l|tr '\n' '\t' >> tax.stat
tail -n+2 tax.txt|cut -f ${i}|grep '__$'|wc -l >> tax.stat; done
cat tax.stat
tail -n+2 tax.txt|cut -f3|sort|uniq -c|awk '{print $2"\t"$1}'|sort -k2,2nr > count.phylum
cat count.phylum
cd ../..
## 5.7 功能注释eggnog/dbcan/arg/antismash
基因注释
mkdir -p temp/prodigal
conda activate eggnog
prodigal -v # V2.6.3
# 50g, 31s, 4m
time tail -n+2 result/metadataS.txt|cut -f1|rush -j 10 \
"prodigal \
-i temp/drep95/dereplicated_genomes/{1}.fa \
-o temp/prodigal/{1}.gff \
-a temp/prodigal/{1}.faa \
-d temp/prodigal/{1}.ffn \
-p single -f gff"
seqkit stat temp/prodigal/*.ffn | sed 's/temp\/prodigal\///;s/\.ffn//;s/[[:blank:]]\{1,\}/\t/g' | cut -f 1,3- \
> result/prodigal.txt
碳水化合物注释
mkdir -p temp/dbcan3 result/dbcan3
time tail -n+2 result/metadataS.txt|cut -f1|rush -j 9 \
"diamond blastp \
--db ${db}/dbcan3/CAZyDB \
--query temp/prodigal/{1}.faa \
--outfmt 6 --threads 8 --quiet --log \
--evalue 1e-102 --max-target-seqs 1 --sensitive \
--block-size 6 --index-chunks 1 \
--out temp/dbcan3/{1}_diamond.f6"
wc -l temp/dbcan3/*.f6|head -n-1|awk '{print $2"\t"$1}'|cut -f3 -d '/'|sed 's/_diamond.f6//'|sed '1 i ID\tCAZy'|less -S > result/dbcan3/gene.count
# format blast2genelist
for i in `tail -n+2 result/metadataS.txt|cut -f1`;do
format_dbcan3list.pl \
-i temp/dbcan3/${i}_diamond.f6 \
-o temp/dbcan3/${i}.list
done
# CAZy type count
for i in `tail -n+2 result/metadataS.txt|cut -f1`;do
tail -n+2 temp/dbcan3/${i}.list|cut -f2|sort|uniq -c|awk '{print $2"\t"$1}'|sed "1 i CAZy\t${i}"|less -S > temp/dbcan3/${i}_CAZy.tsv
done
# merge2table
conda activate humann3
humann_join_tables \
--input temp/dbcan3/ --file_name CAZy \
--output result/dbcan3/cazy.txt
csvtk -t stat result/dbcan3/cazy.txt
# merge to level1
paste <(cut -f1 result/dbcan3/cazy.txt) <(cut -f1 result/dbcan3/cazy.txt|tr '0-9' ' '|sed 's/ //g') | sed '1 s/\tCAZy/\tLevel1/' > result/dbcan3/cazy.L1
summarizeAbundance.py \
-i result/dbcan3/cazy.txt \
-m result/dbcan3/cazy.L1 \
-c 2 -s ',' -n raw --dropkeycolumn \
-o result/dbcan3/sum
# 基因相似度
echo -e 'Name\tCAZy\tIdentity\tGenome' > result/dbcan3/identity.txt
for i in `tail -n+2 result/metadataS.txt|cut -f1`;do
csvtk -t replace -f 2 -p "\d+" -r "" temp/dbcan3/${i}.list | uniq | tail -n+2 | sed "s/$/\t${i}/" >> result/dbcan3/identity.txt
done
csvtk -t stat result/dbcan3/identity.txt
sp_boxplot.sh -f result/dbcan3/identity.txt -m T -F CAZy -d Identity
耐药基因
mkdir -p temp/card result/card
conda activate rgi6
# load database 加载数据库
rgi load -i ${db}/card/card.json \
--card_annotation ${db}/card/card.fasta --local
# Annotation 蛋白注释
# 默认为0, --include_loose 可极大增加结果,519/4657=11.14%; --exclude_nudge结果不变,但jason为空
time for i in `tail -n+2 result/metadataS.txt|cut -f1`;do
# i=X004b2
cut -f 1 -d ' ' temp/prodigal/${i}.faa | sed 's/\*//' > temp/prodigal/protein_${i}.fa
rgi main \
--input_sequence temp/prodigal/protein_${i}.fa \
--output_file temp/card/${i} \
--input_type protein --clean \
--num_threads 8 --alignment_tool DIAMOND > temp/log 2>&1
done
# 附录:常见分析问题和补充代码
## 计算时间统计表
在60核(p)及以上服务器,单样本3个并行推荐16p,混合组装分箱推荐32p。
小数据:2个7.5M样本,在72个核、512GB服务器上测试。
大数据:20个10G样本,在192个核、2TB大内存服务器上测试。
| 步骤 | 数据小(6Gx3) | 数据(10Gx20) | 备注 |
| --- | --- | --- | --- |
| fastqc | 33s | 15m | |
| seqkit | 2s | 15m | |
| fastp | 2s | 40m | |
| kneaddata | 25s | 5h | |
| humann | 34m | 30h | |
| megahit | 39s | 15h | |
| binning | 6h | 16h | -concoct |
| binrefine | 2h | 3h | |
| coverm | 4m | 30m | |
注:m为分,h为小时,d为天
## 补充代码Supplementary scripts
**for循环批量处理样本列表**
# 基于样本元数据提取样本列表命令解析
# 去掉表头
tail -n+2 result/metadata.txt
# 提取第一列样本名
tail -n+2 result/metadata.txt|cut -f1
# 循环处理样本
for i in `tail -n+2 result/metadata.txt|cut -f1`;do echo "Processing "$i; done
# ` 反引号为键盘左上角Esc键下面的按键,一般在数字1的左边,代表运行命令返回结果
## 质控去宿主KneadData
**双端序列质控后是否配对的检查**
双端序列质控后序列数量不一致是肯定出错了。但即使序列数量一致,也可能序列不对。在运行metawrap分箱时会报错。可以kneaddata运行时添加--reorder来尝试解决。以下提供了检查双端序列ID是否配对的比较代码
# 文件
i=C1
seqkit seq -n -i temp/qc/${i}_1_kneaddata_paired_1.fastq|cut -f 1 -d '/' | head > temp/header_${i}_1
seqkit seq -n -i temp/qc/${i}_1_kneaddata_paired_2.fastq|cut -f 1 -d '/' | head > temp/header_${i}_2
cmp temp/header_${i}_?
如果序列双端名称一致,且单样本质控结果异常时使用,适合旧版本:新版全kneaddata 1.12已经将此功能添加至流程,以下代码运行返倒引起错误
序列改名,解决NCBI SRA数据双端ID重名问题,详见[《MPB:随机宏基因组测序数据质量控制和去宿主的分析流程和常见问题》](https://mp.weixin.qq.com/s/ovL4TwalqZvwx5qWb5fsYA)。
# 以0.12.0为例,序列中间不一至存在:1和:2会无结果,
@A01909:80:HFT7YDSX5:2:1101:1289:1000 1:N:0:ATGAATAT+TTAAGTGG
@A01909:80:HFT7YDSX5:2:1101:1289:1000 2:N:0:ATGAATAT+TTAAGTGG
# 以0.12.0为例,最终的结果为无空格,结果/1和/2
@A01909:80:HFT7YDSX5:2:1101:1289:1000.1:N:0:ATGAATAT+TTAAGTGG/1
@A01909:80:HFT7YDSX5:2:1101:1289:1000.1:N:0:ATGAATAT+TTAAGTGG/2
# 以单个样本修改
i=A01
sed -i '1~4 s/ 1:/.1:/;1~4 s/$/\/1/' temp/qc/${i}_1.fastq
sed -i '1~4 s/ 2:/.1:/;1~4 s/$/\/2/' temp/qc/${i}_2.fastq
# 批量修改
sed -i '1~4 s/ 1:/.1:/;1~4 s/$/\/1/' temp/qc/${i}_1.fastq
sed -i '1~4 s/ 1:/.1:/;1~4 s/$/\/1/' temp/qc/${i}_2.fastq
**Perl环境不匹配**
报错'perl binaries are mismatched'的解决
e=~/miniconda3/envs/meta
PERL5LIB=${e}/lib/5.26.2:${e}/lib/5.26.2/x86_64-linux-thread-multi
**Java环境错误**
出现错误 Unrecognized option: -d64,为版本不匹配——重装Java运行环境解决:
conda install -c cyclus java-jdk
若出现错误 Error message returned from Trimmomatic :
Error: Invalid or corrupt jarfile \~/miniconda3/envs/kneaddata/share/trimmomatic/trimmomatic;找不到程序,修改配置文件指定脚本名称
sed -i 's/trimmomatic\*/trimmomatic.jar/' ~/miniconda3/envs/kneaddata/lib/python3.10/site-packages/kneaddata/config.py
**Python环境不匹配-找不到包module**
ModuleNotFoundError: No module named 'importlib.metadata'
找不到包,一般是环境变量错误,先确定是否正常启动conda环境,没有重复启动 conda activate kneaddata。已启动检测环境变量
echo $PATH
# /public/software/env01/bin:/public/home/liuyongxin/miniconda3/envs/kneaddata/bin:/public/home/liuyongxin/miniconda3/condabin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin
确认conda环境是否为第一个路径,此处kneaddata路径前还有更高优先级的目录在前,重设PATH变量,即删除当前conda环境前的所有路径
PATH=/public/home/liuyongxin/miniconda3/envs/kneaddata/bin:/public/home/liuyongxin/miniconda3/condabin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin
## HUMANN物种功能定量
**metaphlan_to_stamp.pl**
# 如果出现下面的错误:
# bash: /db/EasyMicrobiome/script/metaphlan_to_stamp.pl: /usr/bin/perl^M: 解释器错误: 没有那个文件或目录
# sed -i 's/\r//' ${db}/EasyMicrobiome/script/*.pl
**GraPhlAn图**
# metaphlan2 to graphlan
export2graphlan.py --skip_rows 1,2 -i result/metaphlan2/taxonomy.tsv \
--tree temp/merged_abundance.tree.txt \
--annotation temp/merged_abundance.annot.txt \
--most_abundant 1000 --abundance_threshold 20 --least_biomarkers 10 \
--annotations 3,4 --external_annotations 7
# 参数说明见PPT,或运行 export2graphlan.py --help
# graphlan annotation
graphlan_annotate.py --annot temp/merged_abundance.annot.txt \
temp/merged_abundance.tree.txt temp/merged_abundance.xml
# output PDF figure, annoat and legend
graphlan.py temp/merged_abundance.xml result/metaphlan2/graphlan.pdf \
--external_legends
**LEfSe差异分析物种**
* 输入文件:物种丰度表result/metaphlan2/taxonomy.tsv
* 输入文件:样品分组信息 result/metadata.txt
* 中间文件:整合后用于LefSe分析的文件 result/metaphlan2/lefse.txt,这个文件可以提供给www\.ehbio.com/ImageGP 用于在线LefSE分析
* LefSe结果输出:result/metaphlan2/目录下lefse开头和feature开头的文件
前面演示数据仅有2个样本,无法进行差异比较。下面使用result12目录中由12个样本生成的结果表进行演示
# 设置结果目录,自己的数据使用result,演示用result12
result=result12
# 如果没有,请下载演示数据
# wget -c http://www.imeta.science/db/EasyMetagenome/result12.zip
# unzip result12.zip
准备输入文件,修改样本品为组名(可手动修改)
# 预览输出数据
head -n3 $result/metaphlan2/taxonomy.tsv
# 提取样本行,替换为每个样本一行,修改ID为SampleID
head -n1 $result/metaphlan2/taxonomy.tsv|tr '\t' '\n'|sed '1 s/ID/SampleID/' >temp/sampleid
head -n3 temp/sampleid
# 提取SampleID对应的分组Group(假设为metadata.txt中第二列$2),替换换行\n为制表符\t,再把行末制表符\t替换回换行
awk 'BEGIN{OFS=FS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' $result/metadata.txt temp/sampleid|tr '\n' '\t'|sed 's/\t$/\n/' >groupid
cat groupid
# 合并分组和数据(替换表头)
cat groupid <(tail -n+2 $result/metaphlan2/taxonomy.tsv) > $result/metaphlan2/lefse.txt
head -n3 $result/metaphlan2/lefse.txt
方法1. 推荐在线 <https://www.bic.ac.cn/ImageGP/> 中LEfSe一键分析
方法2. LEfSe命令行分析
conda activate lefse
result=result12
# 格式转换为lefse内部格式
lefse-format_input.py $result/metaphlan2/lefse.txt \
temp/input.in -c 1 -o 1000000
# 运行lefse(样本无重复、分组将报错)
run_lefse.py temp/input.in temp/input.res
# 绘制物种树注释差异
lefse-plot_cladogram.py temp/input.res \
$result/metaphlan2/lefse_cladogram.pdf --format pdf
# 绘制所有差异features柱状图
lefse-plot_res.py temp/input.res \
$result/metaphlan2/lefse_res.pdf --format pdf
# 绘制单个features柱状图
# 查看显著差异features,按丰度排序
grep -v '-' temp/input.res | sort -k3,3n
# 手动选择指定feature绘图,如Firmicutes
lefse-plot_features.py -f one --format pdf \
--feature_name "k__Bacteria.p__Firmicutes" \
temp/input.in temp/input.res \
$result/metaphlan2/lefse_Firmicutes.pdf
# 批量绘制所有差异features柱状图
lefse-plot_features.py -f diff \
--archive none --format pdf \
temp/input.in temp/input.res \
$result/metaphlan2/lefse_
**HUMAnN2减少输入文件加速**
HUMAnN2是计算非常耗时的步骤,如果上百个10G+的样本,有时需要几周至几月的分析。以下介绍两种快速完成分析,而且结果变化不大的方法。替换下面for循环为原文中的“双端合并为单个文件”部分代码
方法1. 软件分析不考虑双端信息,只用一端可获得相近结果,且速度提高1倍。链接质控结果左端高质量至合并目录
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
ln -sf `pwd`/temp/hr/${i}_1.fastq temp/concat/${i}.fq
done
方法2. 控制标准样比对时间。测序数据量通常为6~50G,同一样本分析时间可达10h~100h,严重浪费时间而浪费硬盘空间。
可用head对单端分析截取20M序列,即3G,则为80M行
for i in `tail -n+2 result/metadata.txt|cut -f1`;do
head -n80000000 temp/qc/${i}_1_kneaddata_paired_1.fastq > temp/concat/${i}.fq
done
**metaphlan2无法找到数据库**
正常在首次运行时,会自动下载数据库。有时会失败,解决方法:
方法1. 使用软件安装的用户运行一下程序即可下载成功
方法2. 将我们预下载好的数据索引文件,链接到软件安装目录
db=~/db
soft=~/miniconda3
mkdir -p ${soft}/bin/db_v20
ln -s ${db}/metaphlan2/* ${soft}/bin/db_v20/
mkdir -p ${soft}/bin/databases
ln -s ${db}/metaphlan2/* ${soft}/bin/databases/
**CRITICAL ERROR: Can not call software version for bowtie2**
解决问题思路:
查看文件位置是否处在conda环境中:`type bowtie2`。如果不在需要手动设置环境变量的顺序,如果位置正确如在(\~/miniconda2/envs/humann2/bin/bowtie2),请往下看;
检测bowtie2运行情况:`bowtie2 -h`,报错`wd.c: loadable library and perl binaries are mismatched (got handshake key 0xde00080, needed 0xed00080)`。 错误原因为Perl库版本错误,检查Perl库位置:`echo $PERL5LIB`,错误原因没有指向环境,并手动修改perl库位置
# 设置你环境变量位置,最好用绝对路径
e=~/miniconda2/envs/humann2
PERL5LIB=${e}/lib/5.26.2:${e}/lib/5.26.2/x86_64-linux-thread-multi
**metaphlan_hclust_heatmap.py报错AttributeError: Unknown property axisbg**
在网上搜索,axisbg和axis_bgcolor为过时的函数,新版为facecolor,修改为新名称即可 (参考:<https://blog.csdn.net/qq_41185868/article/details/81842971>)
# 定位文件绝对路径
file=`type metaphlan_hclust_heatmap.py|cut -f 2 -d '('|sed 's/)//'`
# 替换函数名称为新版
sed -i 's/axisbg/facecolor/g' $file
**metaphlan2-共有或特有物种网络图**
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {for(i=9;i<=NF;i++) a[i]=$i; print "Tax\tGroup"} \
else {for(i=9;i<=NF;i++) if($i>0.05) print "Tax_"FNR, a[i];}}' \
result/metaphlan2/taxonomy.spf > result/metaphlan2/taxonomy_highabundance.tsv
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {print "Tax\tGrpcombine";} else a[$1]=a[$1]==""?$2:a[$1]$2;}END{for(i in a) print i,a[i]}' \
result/metaphlan2/taxonomy_highabundance.tsv > result/metaphlan2/taxonomy_group.tsv
cut -f 2 result/metaphlan2/taxonomy_group.tsv | tail -n +2 | sort -u >group
for i in `cat group`; do printf "#%02x%02x%02x\n" $((RANDOM%256)) $((RANDOM%256)) $((RANDOM%256)); done >colorcode
paste group colorcode >group_colorcode
awk 'BEGIN{OFS=FS="\t"}ARGIND==1{a[$1]=$2;}ARGIND==2{if(FNR==1) {print $0, "Grpcombinecolor"} else print $0,a[$2]}' \
group_colorcode result/metaphlan2/taxonomy_group.tsv > result/metaphlan2/taxonomy_group2.tsv
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {print "Tax",$1,$2,$3,$4, $5, $6, $7, $8 } else print "Tax_"FNR, $1,$2,$3,$4, $5,$6, $7, $8}' \
result/metaphlan2/taxonomy.spf > result/metaphlan2/taxonomy_anno.tsv
## 生物标志鉴定LEfSe
**lefse-plot_cladogram.py:Unknown property axis_bgcolor**
若出现错误 Unknown property axis\_bgcolor,则修改`lefse-plot_cladogram.py`里的`ax_bgcolor`替换成`facecolor`即可。
# 查看脚本位置,然后使用RStudio或Vim修改
type lefse-plot_cladogram.py
## 物种分类Kraken2
**合并样本为表格combine_mpa.py**
krakentools中combine_mpa.py,需手动安装脚本,且结果还需调整样本名
combine_mpa.py \
-i `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/kraken2\//;s/$/.mpa/'|tr '\n' ' '` \
-o temp/kraken2/combined_mpa
**序列筛选/去宿主extract_kraken_reads.py**
提取非植物33090和动物(人)33208序列、选择细菌2和古菌2157
mkdir -p temp/kraken2_qc
parallel -j 3 \
"/db/script/extract_kraken_reads.py \
-k temp/kraken2/{1}.output \
-r temp/kraken2/{1}.report \
-1 temp/qc/{1}_1_kneaddata_paired_1.fastq \
-2 temp/qc/{1}_1_kneaddata_paired_2.fastq \
-t 33090 33208 --include-children --exclude \
--max 20000000 --fastq-output \
-o temp/kraken2_qc/{1}_1.fq \
-o2 temp/kraken2_qc/{1}_2.fq" \
::: `tail -n+2 result/metadata.txt|cut -f1`
## 组装Megahit
**序列长度筛选**
megahit默认>200,可选 > 500 / 1000 bp,并统计前后变化;如此处筛选 > 500 bp,序列从15万变为3.5万条,总长度从7M下降到3M
mv temp/megahit/final.contigs.fa temp/megahit/raw.contigs.fa
seqkit seq -m 500 temp/megahit/raw.contigs.fa > temp/megahit/final.contigs.fa
seqkit stat temp/megahit/raw.contigs.fa
seqkit stat temp/megahit/final.contigs.fa
**数据太大导致程序中断**
报错信息:126 - Too many vertices in the unitig graph (8403694648 >= 4294967294), you may increase the kmer size to remove tons
解决方法:需要增加k-mer,如最小k-mer改为29,不行继续增加或将数据分批次组装
添加参数: --k-min 29 --k-max 141 --k-step 20
## 二三代混合组装
**metaspades**
# 3G数据,耗时3h
i=SampleA
time metaspades.py -t 48 -m 500 \
-1 seq/${i}_1.fastq -2 seq/${i}L_2.fastq \
--nanopore seq/${i}.fastq \
-o temp/metaspades_${i}
**二三代混合组装OPERA-MS**
结果卡在第9步polishing,可添加--no-polishing参数跳过此步;短序列只支持成对文件,多个文件需要cat合并
perl ../OPERA-MS.pl \
--short-read1 R1.fastq.gz \
--short-read2 R2.fastq.gz \
--long-read long_read.fastq \
--no-ref-clustering \
--num-processors 32 \
--out-dir RESULTS
**二代组装+三代优化**
perl ~/soft/OPERA-MS/OPERA-MS.pl \
--contig-file temp/megahit/final.contigs.fa \
--short-read1 R1.fastq.gz \
--short-read2 R2.fastq.gz \
--long-read long_read.fastq \
--num-processors 32 \
--no-ref-clustering \
--no-strain-clustering \
--no-polishing \
--out-dir temp/opera
结果可用quast或seqkit stat统计对二代组装的改进效果
## 基因序列prodigal
**序列拆分并行预测基因**
(可选)以上注释大约1小时完成1M个基因的预测。加速可将contigs拆分,并行基因预测后再合并。
# 拆分contigs,按1M条每个文件
n=10000
seqkit split result/megahit/final.contigs.fa -s $n
# 生成拆分文件序列列表
ls result/megahit/final.contigs.fa.split/final.contigs.part_*.fa|cut -f 2 -d '_'|cut -f 1 -d '.' \
> temp/split.list
# 9线程并行基因预测,此步只用单线程且读写强度不大
time parallel -j 9 \
"prodigal -i result/megahit/final.contigs.fa.split/final.contigs.part_{}.fa \
-d temp/gene{}.fa \
-o temp/gene{}.gff -p meta -f gff \
> temp/gene{}.log 2>&1 " \
::: `cat temp/split.list`
# 合并预测基因和gff注释文件
cat temp/gene*.fa > temp/prodigal/gene.fa
cat temp/gene*.gff > temp/prodigal/gene.gff
## 基因去冗余cd-hit
**两批基因合并cd-hit-est-2d**
cd-hit-est-2d 两批次构建非冗余基因集
A和B基因集,分别有M和N个非冗余基因,两批数据合并后用cd-hit-est去冗余,计算量是(M + N) X (M + N -1)
cd-hit-est-2d比较,只有M X N的计算量
# 计算B中特有的基因
cd-hit-est-2d -i A.fa -i2 B.fa -o B.uni.fa \
-aS 0.9 -c 0.95 -G 0 -g 0 \
-T 96 -M 0 -d 0
# 合并为非冗余基因集
cat A.fa B.uni.fa > NR.fa
**cd-hit合并多批基因salmon索引时提示ID重复**
# [error] In FixFasta, two references with the same name but different sequences: k141_2390219_1. We require that all input records have a unique name up to the first whitespace (or user-provided separator) character.
# 错误解决
mv temp/NRgene/gene.fa temp/NRgene/gene.fa.bak
# 15G,2m,4G
seqkit rename temp/NRgene/gene.fa.bak -o temp/NRgene/gene.fa
## 基因定量salmon
**找不到库文件liblzma.so.0**
* 报错信息:error while loading shared libraries: liblzma.so.0
* 问题描述:直接运行salmon报告,显示找不到lib库,
* 解决方法:可使用程序完整路径解决问题,`alias salmon="${soft}/envs/metagenome_env/share/salmon/bin/salmon"`
## 基因功能数据库
**综合功能注释KEGG描述整理**
脚本位于 /db/script 目录,<https://www.kegg.jp/kegg-bin/show_brite?ko00001.keg> 下载htext,即为最新输入文件 ko00001.keg
kegg_ko00001_htext2tsv.pl -i ko00001.keg -o ko00001.tsv
# 原核蛋白数据库(需付费购买)建索引
conda activate eggnog
diamond --version # 2.0.13
cd genes/fasta
# 3m, 2G
memusg -t diamond makedb --in prokaryotes.pep.gz \
--db prokaryotes.pep
# ID转换为KO
zless prokaryotes.dat.gz|cut -f1,2|sed "1i Kgene\tKO">prokaryotes.gene2KO
**抗生素抗性CARD**
# 使用3.1.0和3.1.2均有警告,修改序列名至纯字母数数字也无效
# WARNING 2021-07-08 08:58:00,478 : Exception : <class 'KeyError'> -> '5141' -> Model(1692) missing in database. Please generate new database.
# WARNING 2021-07-08 08:58:00,478 : Exception : <class 'KeyError'> -> '5141' -> Model(1692)
# WARNING 2021-07-08 08:58:00,479 : tetM ---> hsp.bits: 60.8 <class 'float'> ? <class 'str'>
**抗生素抗性ResFam**
数据库:<http://www.dantaslab.org/resfams>
参考文献:<http://doi.org/10.1038/ismej.2014.106>
mkdir -p temp/resfam result/resfam
# 比对至抗生素数据库 1m
time diamond blastp \
--db ${db}/resfam/Resfams-proteins \
--query result/NR/protein.fa \
--threads 9 --outfmt 6 --sensitive \
-e 1e-5 --max-target-seqs 1 --quiet \
--out temp/resfam/gene_diamond.f6
# 提取基因对应抗性基因列表
cut -f 1,2 temp/resfam/gene_diamond.f6 | uniq | \
sed '1 i Name\tResGeneID' > temp/resfam/gene_fam.list
# 统计注释基因的比例, 488/19182=2.5%
wc -l temp/resfam/gene_fam.list result/salmon/gene.count
# 按列表累计丰度
summarizeAbundance.py \
-i result/salmon/gene.TPM \
-m temp/resfam/gene_fam.list \
-c 2 -s ',' -n raw \
-o result/resfam/TPM
# 结果中添加FAM注释,spf格式用于stamp分析
awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$4"\t"$3"\t"$2} NR>FNR{print a[$1],$0}' \
${db}/resfam/Resfams-proteins_class.tsv result/resfam/TPM.ResGeneID.raw.txt \
> result/resfam/TPM.ResGeneID.raw.spf
## 分箱MetaWRAP
**Bin refined报错CheckM**
Something went wrong with running CheckM. Exiting...
重新运行1次,错误仍在;
删除concoct输入,尝试提纯
**MetaWRAP分箱注释Bin classify & annotate**
# Taxator-tk对每条contig物种注释,再估计bin整体的物种,11m (用时66 min)
metawrap classify_bins -b temp/bin_refinement/metawrap_50_10_bins \
-o temp/bin_classify -t 3 &
# 注释结果见`temp/bin_classify/bin_taxonomy.tab`
# export LD_LIBRARY_PATH=/conda2/envs/metagenome_env/lib/:${LD_LIBRARY_PATH}
# 这是动态链接库找不到时的一个简单的应急策略
#ln -s /conda2/envs/metagenome_env/lib/libssl.so.1.0.0 .
#ln -s /conda2/envs/metagenome_env/lib/libcrypto.so.1.0.0 .
# 基于prokka基因注释,4m
metaWRAP annotate_bins -o temp/bin_annotate \
-b temp/bin_refinement/metawrap_50_10_bins -t 5
# 每个bin基因注释的gff文件bin_funct_annotations,
# 核酸ffn文件bin_untranslated_genes,
# 蛋白faa文件bin_translated_genes
ls -sh temp/bin_annotate/prokka_out/bin.1/
**分箱定量Bin quantify**
# 耗时3m,系统用时10m,此处可设置线程,但salmon仍调用全部资源
metawrap quant_bins -b temp/bin_refinement/metawrap_50_10_bins -t 4 \
-o temp/bin_quant -a temp/megahit/final.contigs.fa temp/qc/ERR*.fastq
# 文件名字改变
# 结果包括bin丰度热图`temp/bin_quant/bin_abundance_heatmap.png`
# 原始数据位于`temp/bin_quant/bin_abundance_table.tab`
ls -l temp/bin_quant/bin_abundance_heatmap.png
**GTDB的文件名不存在错**
# ERROR: ['BMN5'] are not present in the input list of genome to process,但并无此菌,可能是名称 中存在"-"或".",替换为i
# 修改metadata
sed 's/-/i/;s/\./i/' result/metadatab.txt > result/metadata.txt
# 修改文件名
awk 'BEGIN{OFS=FS="\t"}{system("mv temp/antismash/"$1".fna temp/antismash/"$2".fna")ll }' <(paste result/metadatab.txt result/metadata.txt|tail -n+2)
# 版本更新记录
**1.08 2020.7.20**
1. KneadData提供数据预处理双端标签唯一命令,兼容最新版;
2. 提供HUMAnN3测试版的安装和分析流程(附录1);
3. eggNOG升级为emapper 2.0和eggNOG 5.0流程,结果列表从13列变为22列,新增CAZy注释。emapper 1.0版本见附录2。
**1.09 2020.10.16**
1. 新增二、三代混合组装OPERA-MS软件使用 (31Megahit)
2. 新增eggNOG-mapper结果COG/KO/CAZy整理脚本summarizeAbundance.py,删除旧版Shell+R代码 (32Annotation)
3. 新增MetaWRAP单样本分箱流程 (33Binning)
4. 新增dRep实现基因组去冗余 (34Genomes)
5. 新增GTDB-Tk基因组物种注释和进化树构建 (34Genomes)
**1.10 2021.1.22**
1. 增加删除中间文件部分,节约空间,防止硬盘写满;
2. 正文的补充分析方法、常见问题移至附录,按软件名、问题/方法分级索引;
3. 软件使用前,增加检查软件版本命令,方便文章方法中撰写准确版本;
4. 删除不稳定的humann3、过时的eggnog版本教程;
5. 增加kraken2新环境, 增加bracken, krakentools新工具;
6. kraken2结果新增beta多样性PCoA,物种组成堆叠柱状图;
7. 增metaspades二、三代组装代码示例;
8. 新增KEGG层级注释整理代码;
9. 更新dbcan3中2018版为2020版;
10. 新增CARD本地分析流程;
**1.11 2021.5.7**
1. 增加prodigal基因预测并行版方法,使用seqkit split拆分后并行,数10倍加速单线程基因预测步骤;
2. 增加megahit拼装结果片段大小选择步骤,使用seqkit -m按长度筛选,并统计筛选前后变化;
3. 不常用或可选代码调整到附录
4. 两批数据快速合并去冗余cd-hit-est-2d
5. 二三代混合组装OPERA-MS的混装和3代优化代码
**1.12 2021.8.20**
1. 新增并行管理软件rush,比parallel更易安装,绿色版无依赖关系,整合在db/linux/目录中
2. 新增seqkit,可以统计序列数据量,支持序列长度过滤,格式转换等;
3. 新增质控软件fastp,软件fastqc更快,适合单独质控不去宿主;
4. kraken2新数据库,同样大小下注释率提高明显;
5. eggNOG软件和数据库配套升级
6. GTDB-tk软件和数据库需要配套重新才可使用新版25万基因组数据库
**1.13 2021.11.19**
1. 陈同参与EasyMicrobiome的更新,并提交了mac版本代码
2. 新增humann2运行bowtie2出错的解决方案
3. 新增软件conda环境下载安装方式,且作为首选
4. 新增kneaddata自定义物种基因组数据库示例
**1.14 2022.3.25**
1. EasyMicrobiome升级为1.14
2. 升级miniconda2为miniconda3
3. dbcan3从2020/7/31的808M更新为2021/9/24版1016M,格式变化,配套format_dbcan3list.pl更新
4. 新增eggnog环境,包含emapper 2.1.6,summarizeAbundance.py含pandas (conda install sklearn-pandas),配套更新数据库
5. rgi更新到最新版及配套代码
**1.15 2022.5.27**
1. 陈同老师全面更新课程,并在新服务器上重新布置所有软件和数据库
2. 课题尝试改为长期:自学理论课程视频,每周线上答疑,持续2个月完成实操
**1.18 2023.4.7**
1. 课程恢复为3天连续学习模式
2. 更新所有软件和数据库为可成功安装的最新版
3. 更新软件和数据备份至微生物所和百度网盘
**1.19 2023.7.13**
1. HUMAnN2+MetaPhlAn2为3和4
2. Kraken2数据库不同版本统一官方名称,仍使用3月版本数据库,最新版6月数据库官方文件有不完整
3. GTDB-tk数据库更新为214版
**1.20 2023.11.24**
1. MetaPhlAn4新增物种注释转换为GTDB、多样性计算脚本
2. 整合陈同老师用Pipeline的修正
3. CoverM定量MAGs相对丰度、结果合并和求均值,并添加到进入树注释结果中
4. drep的conda包为3.4.2缺少checkm(267M),替换为旧版2.6.2(526M)
5. dbCAN3数据库更新为2023版,diamond新版建索引更快
6. Kneaddata质控跳过,fastp质控为必选步骤
7. mutliqc升级1.14为1.15
8. 增加第五章:单菌基因组分析流程
9. 更新Kraken2数据库为20231009版本,新增alpha, beta多样性、Krona网页、Pavian桑基图
10. 新增可选的checkm2评估
**正在开发中功能**
1. rgi应用于菌群分析及结果展示
2. antisamsh应用于菌群分析及结果展示
3. cazy应用于菌群分析及结果展示