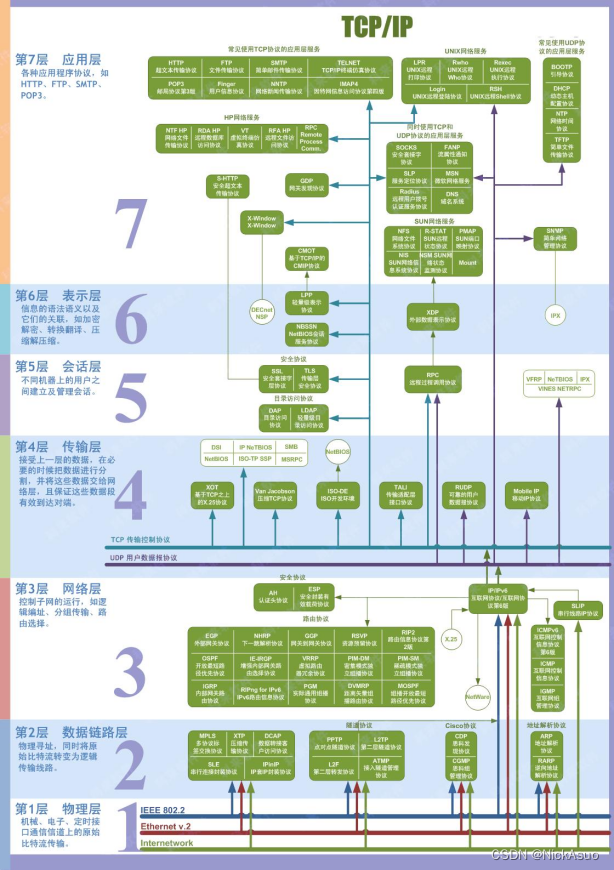
今天给同学们分享一篇鞘脂相关基因在乳腺癌临床中的作用的生信文章“Exploring the role of sphingolipid-related genes in clinical outcomes of breast cancer”,这篇文章于2023年2月14日发表在Front Immunol期刊上,影响因子为8.786。尽管癌症研究取得了巨大进步,但乳腺癌(BC)仍然是一个主要的健康问题,并且是影响全球女性的最常见癌症。乳腺癌是一种高度异质性的癌症,具有潜在的侵袭性和复杂的生物学特性,针对特定亚型的精准治疗可能会提高乳腺癌患者的生存率。鞘脂是脂质的重要组成部分,在肿瘤细胞的生长和死亡中起着关键作用,并且越来越成为新的抗癌疗法的主题。鞘脂代谢(SM)的关键酶和中间体在调节肿瘤细胞和进一步影响临床预后方面发挥着重要作用。
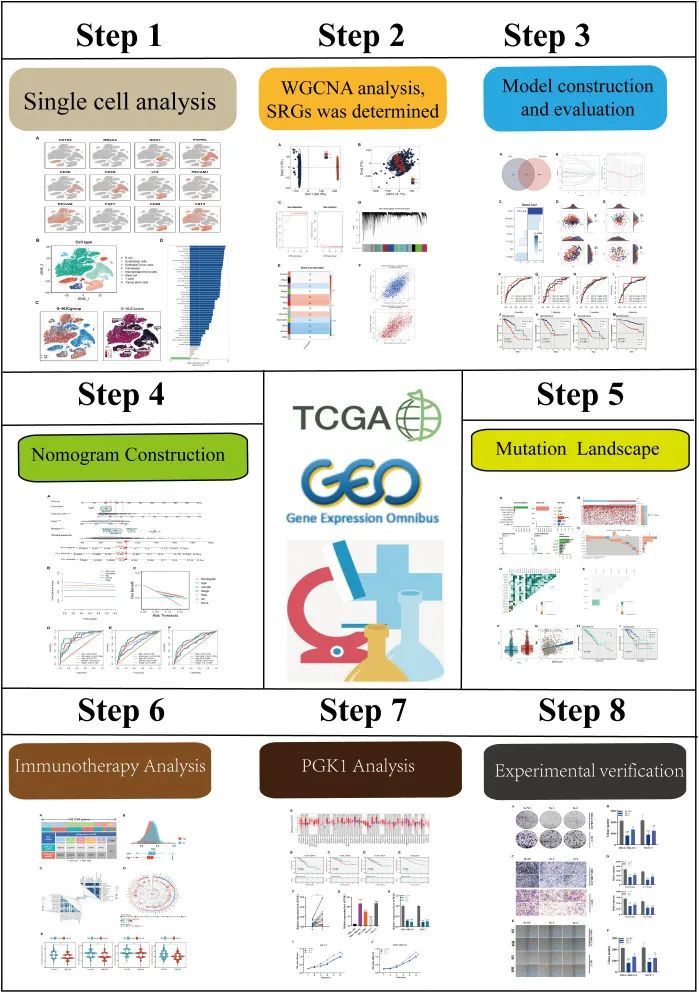
图1 研究流程图
1. 单细胞测序数据分析
在单细胞数据集上,作者进行了质量控制。为了确认细胞样本的有效性,作者去除了一些细胞并限制了线粒体基因、核糖体基因和红细胞基因的百分比。测序深度和细胞内总序列表现出显着的正相关性。PCA还原图未显示细胞周期有任何明显变化。研究包含10个样本,其中每个样本的细胞分布基本保持不变。这表明对样品没有明显的批次影响,可用于进一步分析。随后,所有细胞都通过降维算法进行分类,即t-SNE分为19个更详细的簇(图2A)。有八种不同类型的细胞,例如成纤维细胞、单核细胞/巨噬细胞和肿瘤细胞(图2B)。AUCellR包用于确定每个细胞的SM活动,以探索SRG的表达特征。在表达更多基因的细胞中观察到更高的AUG值,这些细胞主要是橙色B细胞和浆细胞(图2C、D)。所有细胞都被分配了相应SRG的AUC分数,并根据AUC分数阈值分为两组(高鞘脂AUC组和低鞘脂AUC组)。为了阐明不同AUC分数的潜在生物学机制,作者进行了差异分析和功能分析,以确定与高和低鞘脂-AUC亚组之间的糖基化相关的DEG和途径。作者通过差异分析确定了1221个最有可能影响SM的基因。这些通路主要与氧化磷酸化、细胞凋亡、脂肪酸代谢和p53通路有关(图2E)。
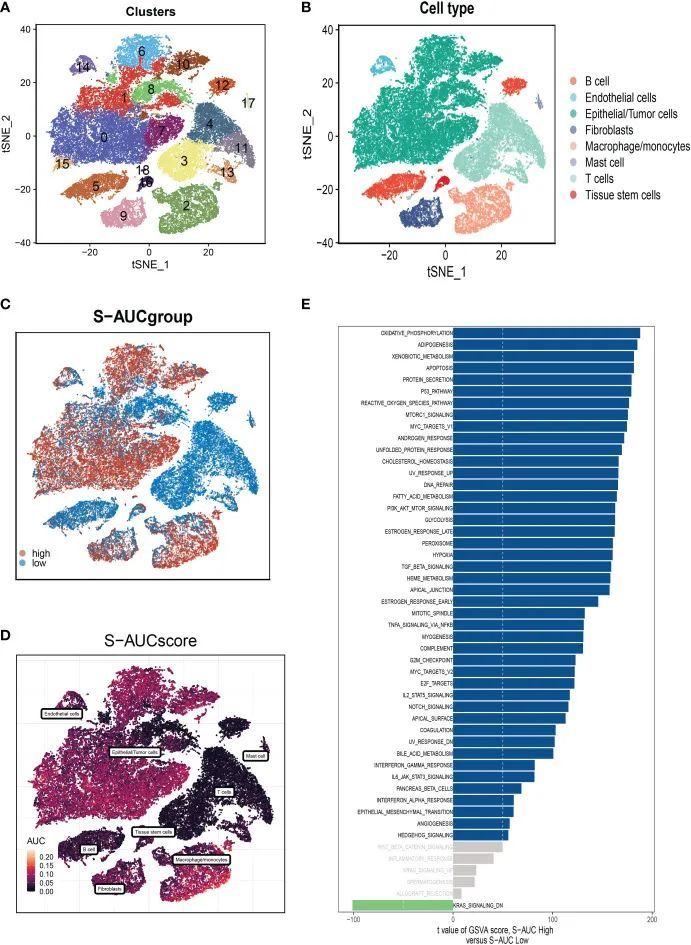
图2 细胞亚群的注释和差异表达基因的鉴定
2. WGCNA分析
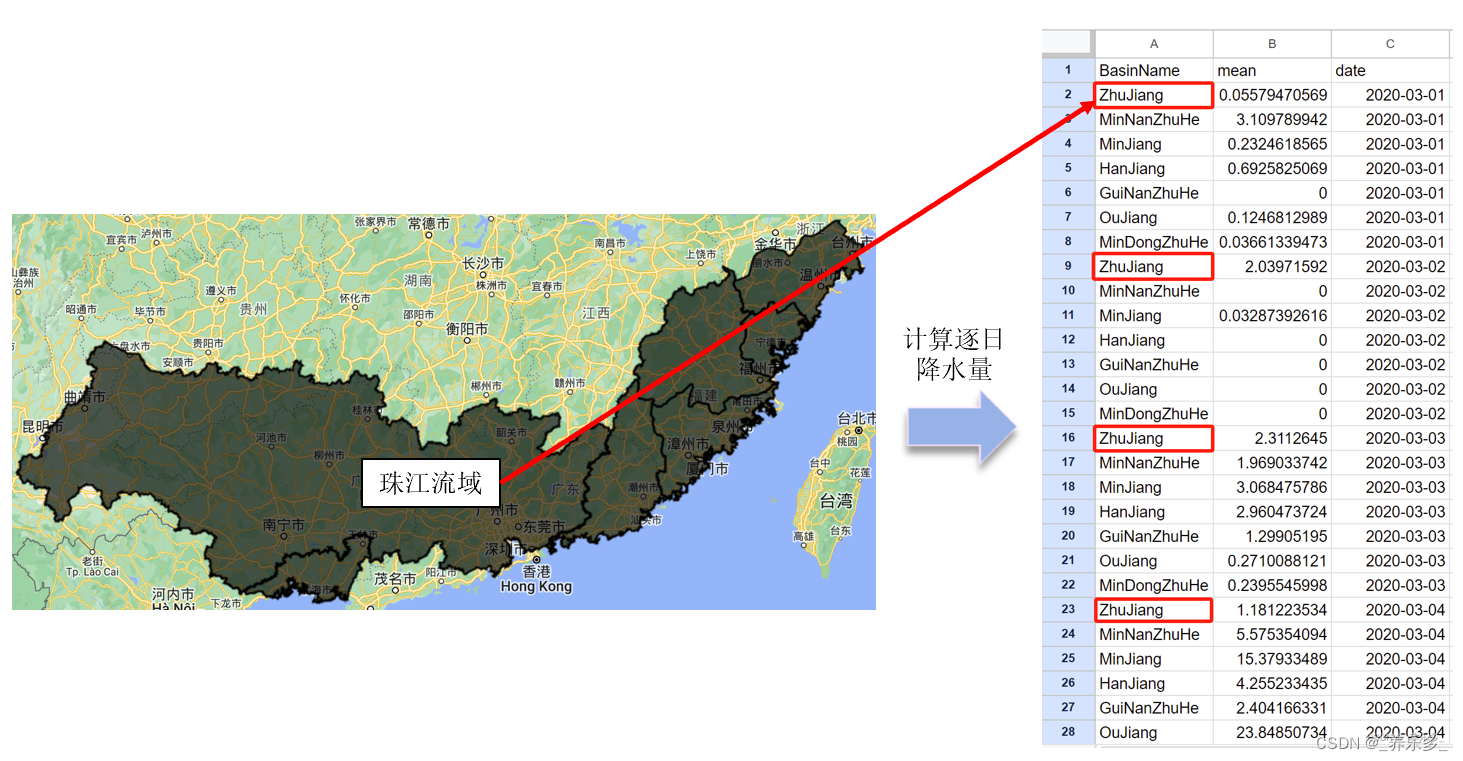
图3A显示TCGA和GEO队列独立,具有显着的批次效应。去除批次效应后,获得了更准确的结果(图3B)。WGCNA用于更详细地寻找与鞘脂共变的基因组。如图3C所示,当软域值为6时,数据更符合幂律分布,平均连通性趋于稳定;使数据适合进一步研究。如图3D所示,将相似度小于0.25的模块合并后,将最小模块数设置为100,将deepSplit设置为2后,生成了12个非灰色模块。根据图3E、F,作者发现蓝色和棕色模块,每个模块包含3,787个基因,与SM的关系最密切。
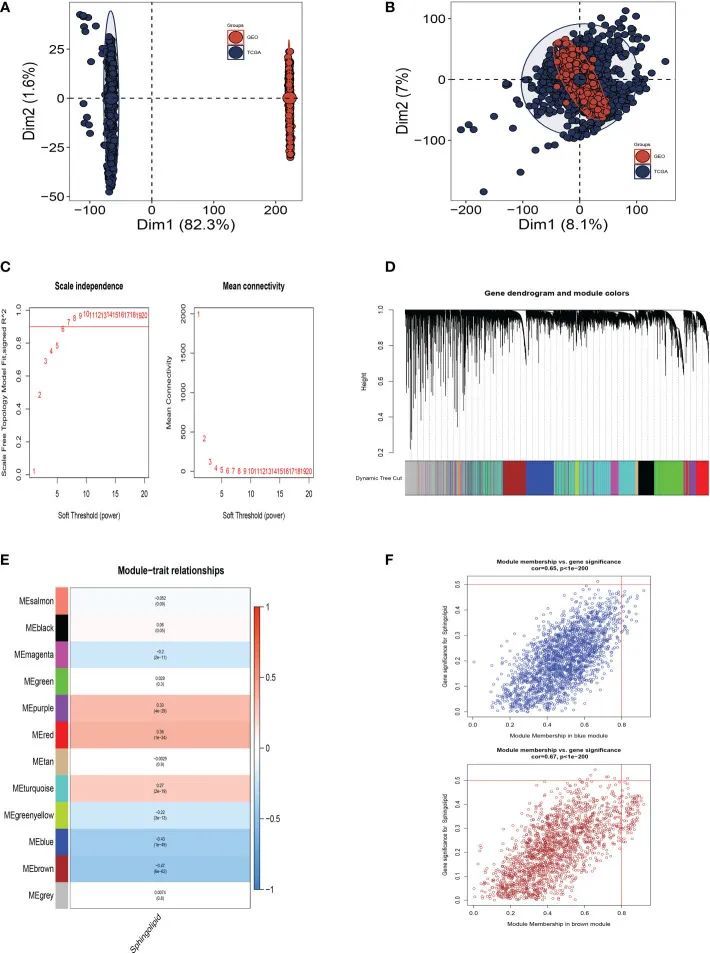
图3 WGCNA分析
3. 鞘脂相关预后模型的构建与验证
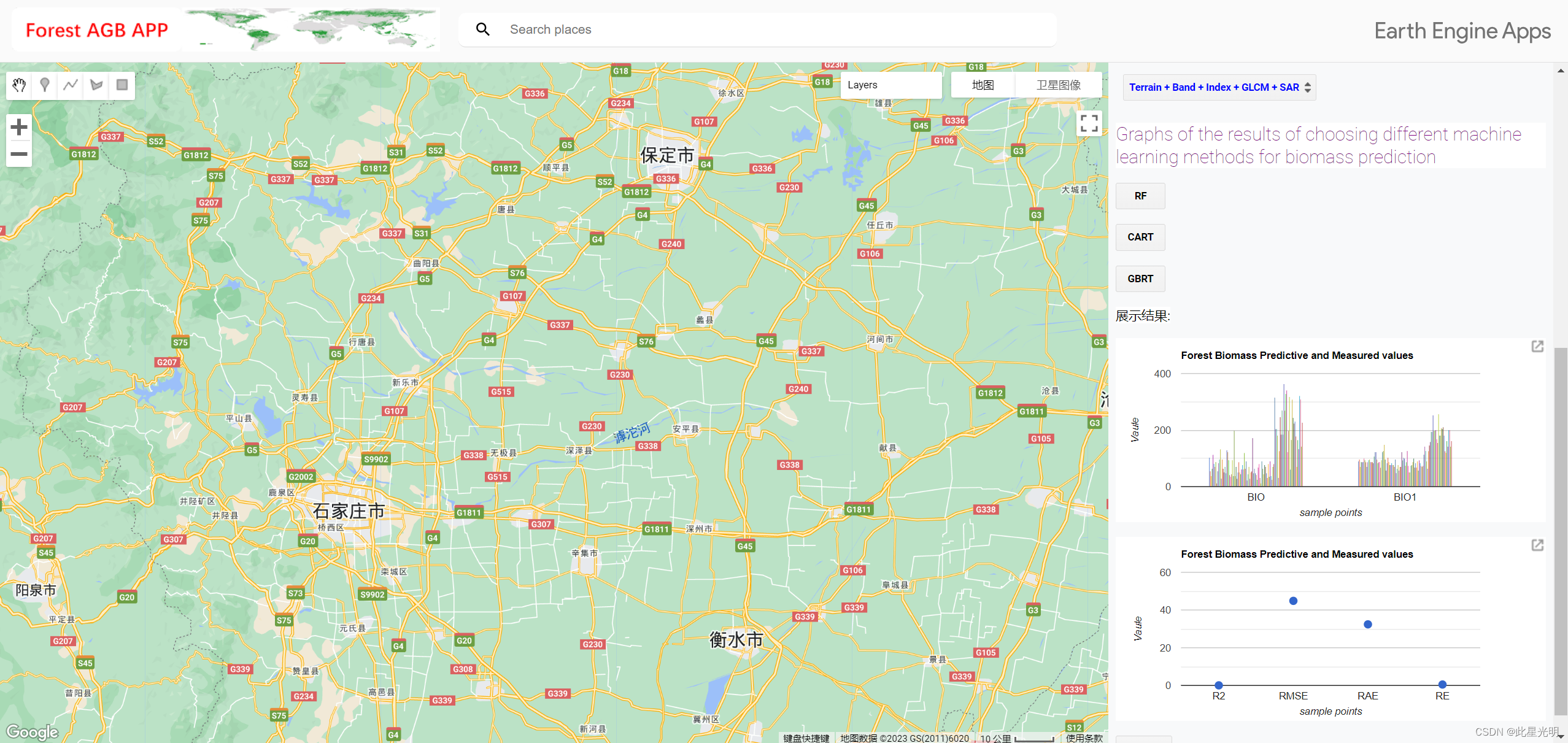
为了进一步探讨SRGs如何与BC患者的预后相关,作者对影响单细胞中获得的鞘脂代谢活性的最相关基因取交集。此外,Bulk-RNA分析和303个基因用于后续分析(图4A)。作者使用TCGA-BC中的训练集进行模型构建,单因素分析共获得63个预后基因。接下来,采用LASSO-Cox回归分析来开发预后模型(图4B)。在最佳正则化参数下最终筛选出7个模型基因(TAGLN2、CHI3L1、PGK1、ATP6AP1、MIA、PSME1、TTC39C)。Coefi和Expi分别代表每个模型基因的系数和表达量。根据中值,患者被分为高危组和低危组。在用于构建模型的七个基因中,三个是风险因素,四个是保护因素(图4C)。结果发现,通过分别对训练集和验证集中模型的七个基因进行PCA和t-SNE评估,该模型可以有效地将训练队列和测试队列中的BC患者分组(图4D、E)。作者在训练和测试队列中进行了ROC曲线分析,以进一步研究鞘脂在评估BC患者预后中的精确度。TCGA训练、测试和完整队列的曲线下面积(AUC)值均超过0.7(图4F-H)。作者发现GEO测试组在1、3和5年时的曲线下面积分别为0.768、0.707和0.711(图4I)。在图4J–L中,作者发现高风险组在TCGA训练、测试和整个队列中的预后较差。同样,作者发现GSE20685测试队列中高危组患者的预后明显低于高危组患者(图4M)。这表明与鞘脂相关的预后模型在预测两个队列的患者结果方面都非常准确。
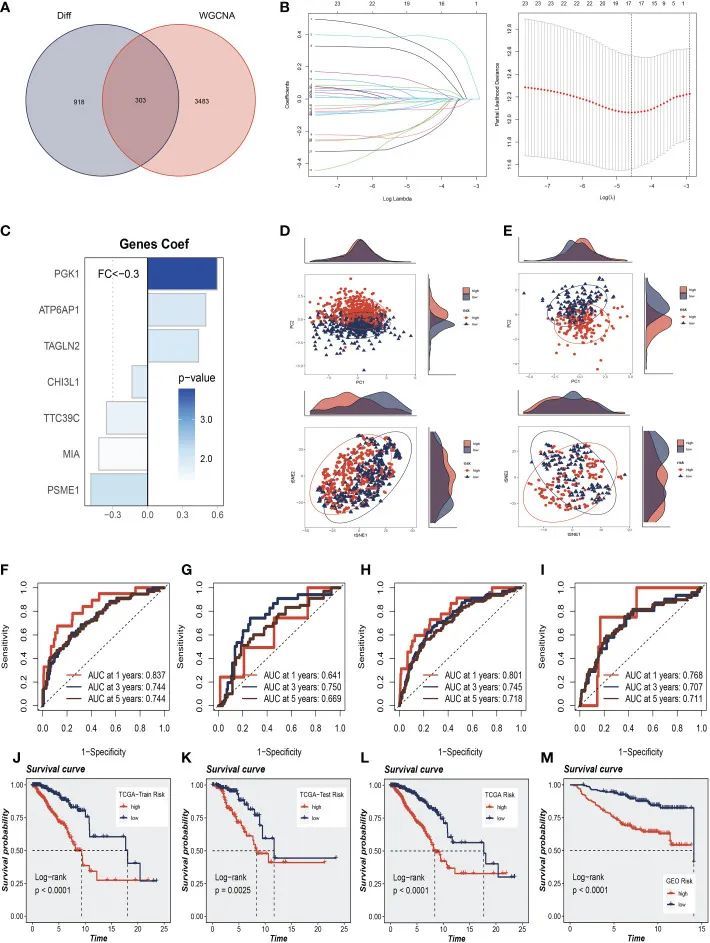
图4 鞘脂相关预后模型的构建与验证
4. 列线图的构建
使用临床信息和风险评分,创建了列线图以更准确地量化BC患者的风险(图5A)。列线图可以帮助更准确地确定患者风险并指导未来的治疗决策。作者还进行了决策曲线和一致性指数研究,确定每个临床特征的面积和横轴,以评估临床决策价值。结果表明,该列线图的功效优于其他临床指标,表明它可有效预测患者的预后,可作为临床决策工具(图5B、C)。进行预后ROC分析以彻底评估该列线图的准确性。根据调查结果,曲线下面积(AUC)在1年、3年和5年时分别为0.888、0.804和0.765(图5D-F)。
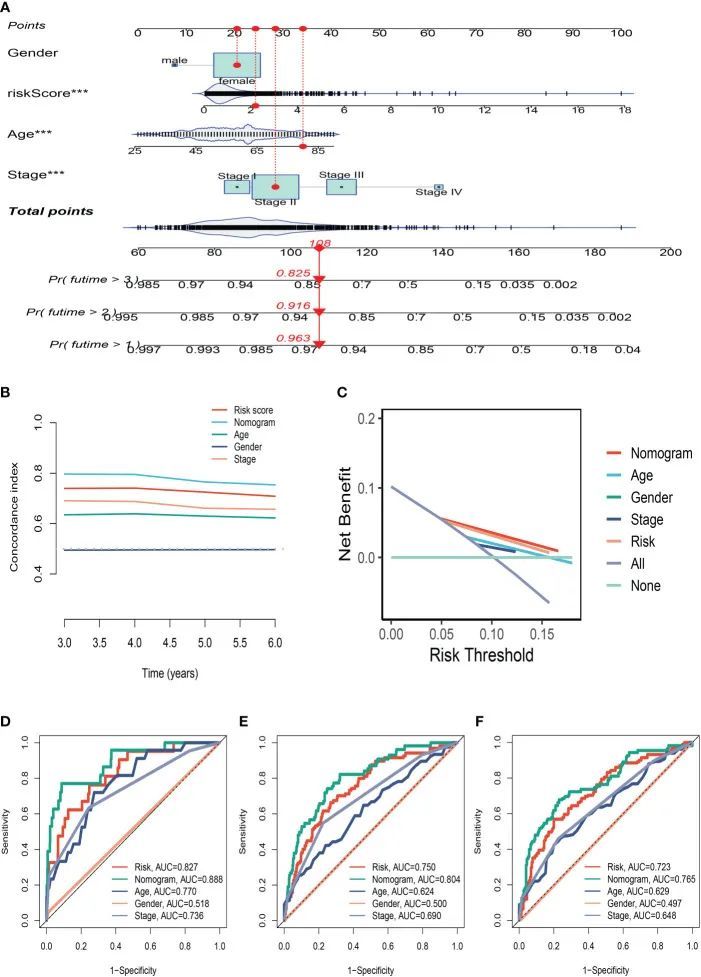
图5 列线图的构建
5. 临床相关性分析
作者制作了临床热图以确定两个风险组之间临床特征的差异。图6A显示了这两组之间在肿瘤年龄、T和N分期方面的显着差异。有趣的是,高危组中有年龄较大的患者和更晚期的N、M期患者(图6B–E)。进一步讨论了两组之间耐药性的差异,并在图6F-I中进行了介绍。作者发现CP724714、Lapatinib、WZ3105和Pyrimethamine可能是治疗高危人群的候选药物。这将为临床选择最适合的药物提供参考。
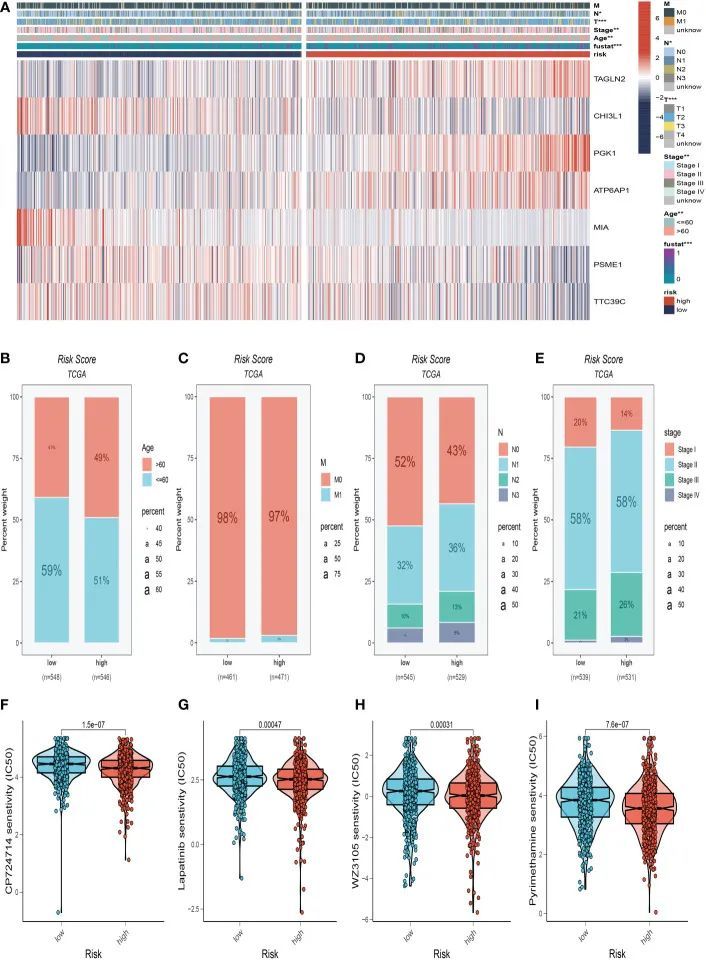
图6 临床相关性分析
6. 突变景观分析
BC样本中的突变概述如图7A所示,其中最常见的突变类型是错义突变。前3个最常见的突变基因是TP53、MUC16和MAP3K1。作者还检查了高风险和低风险组中的代表性基因变异(图7B)。TP53、GATA3、ZFHX4、SPTA1、DMD等基因突变频率在高危人群中位居前五位。低危组突变频率最高的前5个基因分别为PIK3CA、CDH1、MAP3K1、PTEN、NEB。图7C分析了961个BC样本中用于构建模型的7个SRG的突变,其中CHI3L1和PGK1发生了突变。此外,作者检查了前25个基因的突变共生,发现PIK3CA和NEB、MAP3K1、KMT2C、GATA3、CDH1和TP53都共享突变共生(图7D)。此外,作者发现CHI3L1和PGK1之间存在突变共生(图7E)。两个风险组之间的肿瘤突变负荷(TMB)水平存在显着差异,风险评级与TMB值之间存在正相关关系(图7F、G)。作者调查了TMB联合两组对OS的预后影响。生存分析表明,较高的TMB水平与较差的OS相关(图7H、I)。
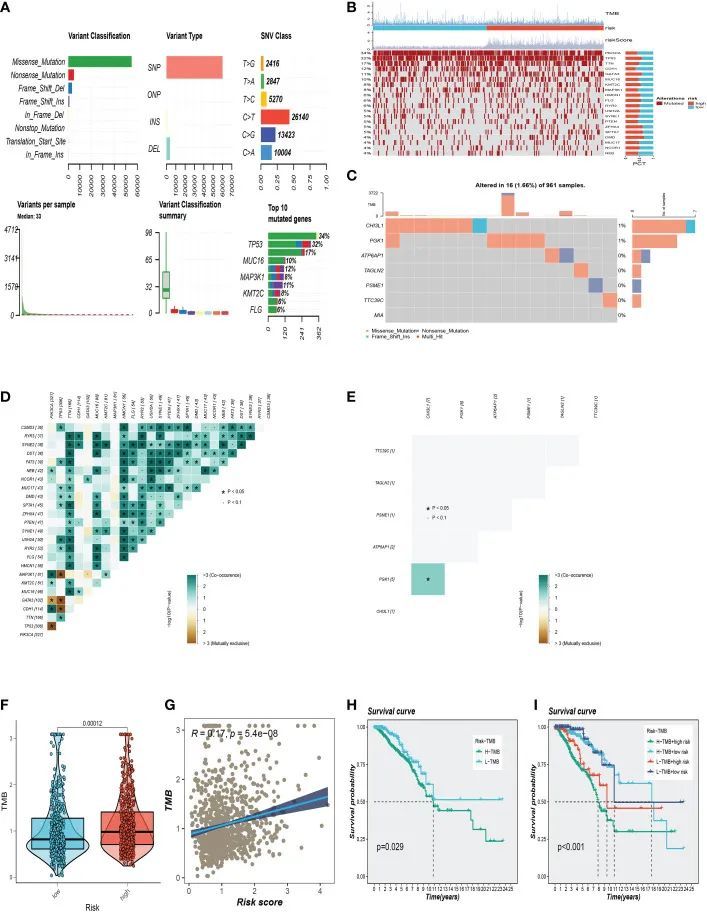
图7 突变分析
7. 免疫景观和免疫治疗
作者使用CIBERSORT方法确定了每个样本中的免疫细胞浸润程度,以更好地了解TCGA-BC队列中22种肿瘤浸润免疫细胞(TIC)相对含量的分布和关联。除了非特征性细胞、常见淋巴祖细胞和M2巨噬细胞外,低风险组的免疫浸润水平似乎高于高风险组(图8A)。然后,低风险组的基质评分、免疫学评分和ESTIMATE评分较高,表明该组TME的总体免疫水平和免疫原性较高。作者还研究了肿瘤纯度,结果显示两者呈正相关(图8B、C)。由于免疫检查点对于免疫疗法在肿瘤中取得成功的重要性,作者还研究了免疫检查点表达在两组之间的差异。37个免疫检查点基因在低风险患者中显着上调。在高风险组中观察到免疫检查点基因CD276和TNFSF4的显着升高(图8D)。患有这种肿瘤亚型的患者可能会受益于针对表达增加的免疫检查点的靶向治疗。
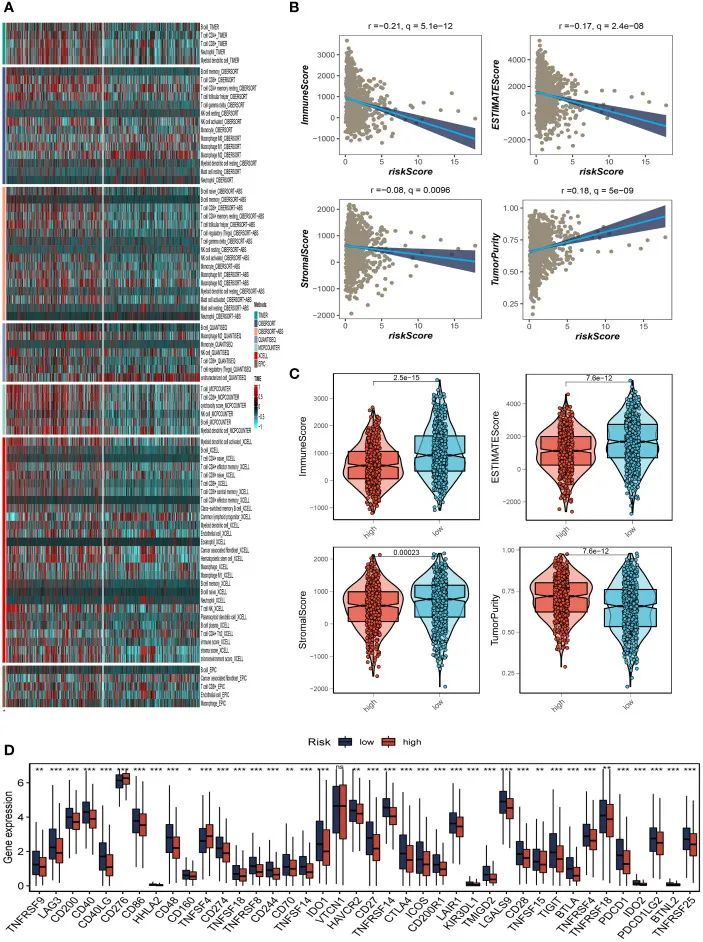
图8 免疫微环境分析
8. SRGs风险评分预测治疗反应评估
作者首先将高风险和低风险人群的免疫分型与传统的免疫分型进行了比较。作者比较了BC在各种风险类别中的免疫亚型分布。研究结果表明,各组的免疫分型差异显着(图9A)。关于TMB和免疫疗法如何相互作用,为了确定具有不同风险模式的患者对免疫疗法的反应是否不同,进行了肿瘤免疫功能障碍和排除(TIDE)分析。根据研究结果,高风险组对免疫疗法的反应更好,因为他们的TIDE评分较低(图9B)。然后进一步研究了SRG风险评分与阳性免疫检查点阻断(ICB)相关信号之间的关系。研究结果表明,风险评分与DNA复制、细胞周期、范可尼贫血途径、同源重组、错配修复和核苷酸切除修复之间存在显着正相关(图9C)。从图9C可以看出,CHI3L1与免疫相关基因呈显着正相关。从前面的分析可以看出CHI3L1的HR为0.87415,由此可以推测CHI3L1可能是激活肿瘤微环境适应性免疫反应的关键基因,ATP6AP1与免疫基因显着负相关。从前面的分析可以看出ATP6AP1的HR为1.64752,因此可以推测ATP6AP1可能对免疫反应具有抑制作用,从而促进肿瘤的生长和转移。从以上分析,作者可以推测CHI3L1和ATP6AP1可以通过改变TME的状态来决定患者的预后,这可能为临床治疗提供新的思路(图9D)。此外,IPS有助于筛查易受免疫疗法影响的患者。在作者的研究中,低风险亚型比高风险亚型具有更高的IPS和阻滞剂评分,突出表明低风险患者可能更容易接受免疫检查点抑制剂(ICI)治疗并获得更显着的益处(图9E)。
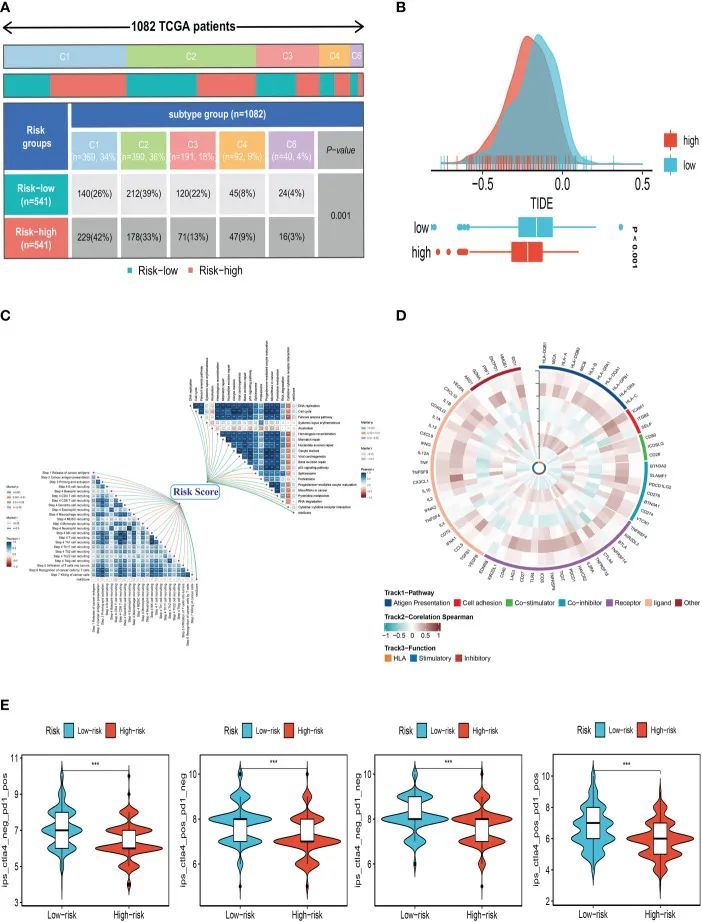
图9 治疗反应相关性分析
9. PGK1在BC样本中的表达和预后
PGK1表达水平的泛癌分析显示,相对于正常组织,PGK1在BC中高表达(图10A)。为了进一步判断PGK1的预后,在TCGA中进行了总生存期(OS)、疾病特异性生存期(DSS)和无进展生存期(PFS)的生存分析,PGK1高表达的患者预后较差预后(图10B–D)。同时,作者深入分析了GSE22219中的PGK1无复发生存期(RFS),并获得了相同的结果(图10E)。同样,GEPIA数据集显示了类似的结果。作者通过体外实验进一步确定了PGK1的功能。作者对来自医院的20对乳腺癌组织样本进行了相同的验证。在临床样本中,作者观察到类似的表达趋势(图10F)。正如预期的那样,PGK1在20对组织样本中的14对中高表达。图10G显示与HBL-100细胞系相比,PGK1在四种BC细胞系中高表达,并且与其他乳腺癌细胞相比,PGK1在MDA-MB-231和MCF-7细胞中的表达最高。这些结果证实了上述生物信息学研究的准确性。
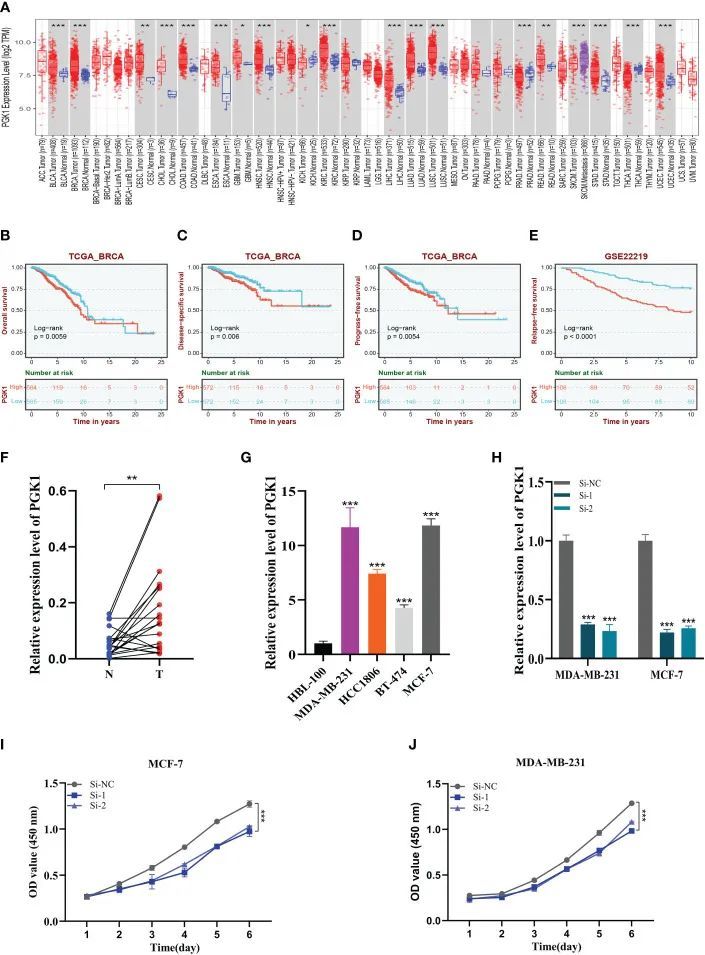
图10 PGK1的表达分析和实验验证
10. PGK1的实验验证
如前所述,PGK1在MDA-MB-231和MCF-7细胞系中的表达水平最高,因此作者对这两种细胞系进行了基因敲除。这两个细胞系的PGK1表达发生了相当大的变化(图10H)。在CCK-8中,作者观察到PGK1敲除MCF-7细胞的增殖活性与对照细胞相比显着降低(图10I)。在细胞系MDA-MB-231中观察到类似的结果(图10J)。为进一步验证PGK1对BC细胞增殖能力的影响,作者还进行了克隆实验。结果显示,在PGK1基因敲低后,两种细胞系形成的集落数量和体积减少(图11A)。接下来,作者通过愈合和transwell实验来分析PGK1对BC细胞迁移和侵袭能力的影响。结果显示,敲低PGK1后,BC细胞的迁移和侵袭能力明显减弱(图11B、C)。如图11D所示,PGK1敲低抑制了肿瘤生长,导致与对照组相比肿瘤体积和重量减少。
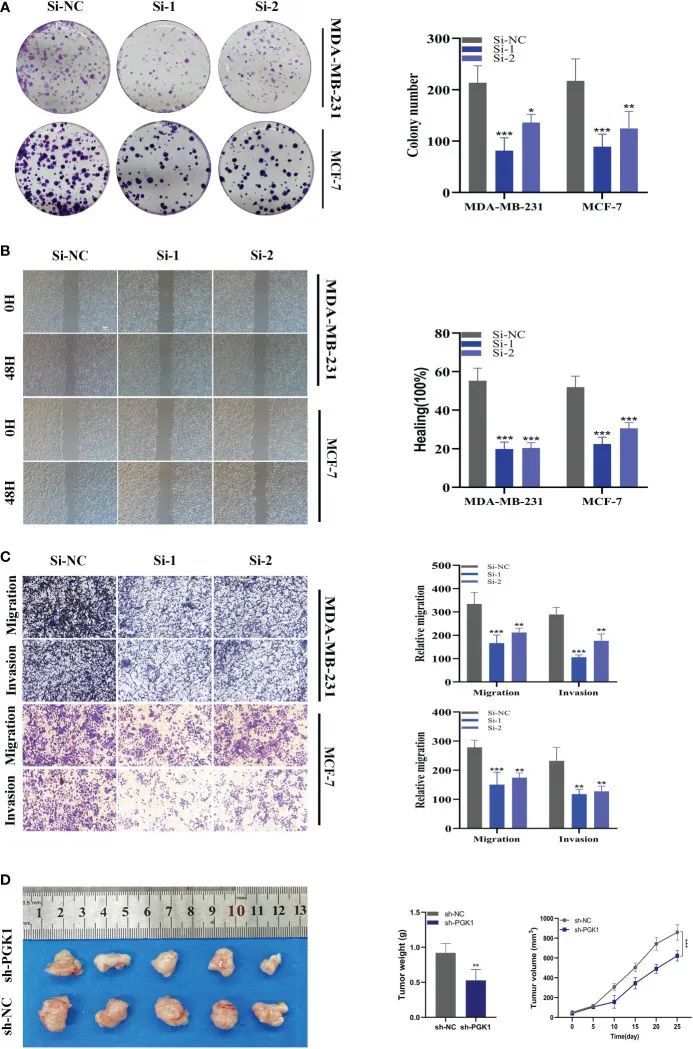
图11 PGK1敲低后的体外实验
总结
总之,作者的结果表明,由7个SRG构建的模型可以很好地预测BC患者的预后。此外,作者通过细胞实验验证了PGK1在BC中的功能,并筛选了BC的候选疫苗基因。这些结果可能会为制定新的BC治疗计划提供有用的信息。