使用hifiasm-meta进行Hifi长读宏基因组序列组装
Metagenome assembly of high-fidelity long reads with hifiasm-meta

Article,2022-05-09
Nature methods, [IF 48]
DOI:https://doi.org/10.1038/s41592-022-01478-3
原文链接:https://www.nature.com/articles/s41592-022-01478-3
第一作者:Xiaowen Feng
通讯作者:Heng Li (李恒)
合作作者:Haoyu Cheng;Daniel Portik
主要单位:
哈佛大学医学院生物医学信息系 (Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA)
Dana-Farber癌症研究所数据中心 (Department of Data Sciences, Dana-Farber Cancer Institute, Boston, MA, USA)
太平洋生物科技公司(Pacific Biosciences, Menlo Park, CA, USA)
- 摘 要 -
宏基因组样本的从头组装是研究微生物群落的常用方法。当前针对短读长或错误率高的长读长开发的宏基因组组装软件尚未对组装准确的长读长序列进行优化。因此,我们开发了hifiasm-meta,一种利用近期出现的高精度的宏基因组数据进行宏基因组组装的软件。通过使用七个经验数据集进行评估,hifiasm-meta在每个数据集重建了数十到数百个完整的闭环细菌基因组,始终优于其他宏基因组组装软件。
专业词汇
reads:读长,就是我们测序产生的短读序列,通常一代和三代的读长读长在几千到几万bp之间,二代的相对较短,平均是几十到几百bp。
contig:重叠群,就是不同读长之间的overlap交叠区,拼接成的序列就是contig。
scaffold: 支架,这是比contig还要长的序列,获得contig之后还需要构建paired-end或者mate-pair库,从而获得一定片段的两端序列,这些序列可以确定contig的顺序关系和位置关系,最后contig按照一定顺序和方向组成scaffold,其中形成scaffold过程中还需要填补contig之间的空缺。
- 主要内容 -
短读长宏基因组组装通常能产生长度为数十千碱基(kbp)的重叠群,约为细菌基因组大小的1%。截至2019年9月,经过多年的宏基因组测序,只有62个完整的细菌基因组从宏基因组样本中组装而成。尽管当前可以使用分箱算法将短的重叠群聚类到宏基因组组装基因组(MAG)中,但是分箱会产生较高的错误率,导致下游分析复杂化或错误。短读长MAG的局限性,激发了metaFlye的开发,唯一已发表的专门用于长读长宏基因组组装的软件。metaFlye是基于Flye开发的,其适用于错误率约10%的嘈杂长读长数据组装,不适用于PacBio产生的高准确度数据组装,并且对于单物种HiFi组装来说是次优的。为了充分利用长而精确的HiFi读长的全部优势,我们开发了hifiasm-meta,将作者早期开发的hifiasm应用到宏基因组样本组装中。
与单个物种的组装相比,宏基因组组装带来了几个独特的挑战,例如PacBio HiFi数据中读长长度差异较大,以及某些单倍型的高倍性与低覆盖率相结合。作者在hifiasm-meta中做了几个重大改变来应对这些挑战。首先,hifiasm-meta具有读长选择步骤,可以减少高丰度菌株的覆盖率,而不会丢失低丰度菌株的数据。其次,在组装图的构建过程中,hifiasm-meta试图保护低覆盖率基因组中的序列,这些序列可能被视为嵌合序列并被原始hifiasm丢弃。第三,hifiasm-meta 只有在推断出与读长完全重叠的其他序列来自同一单倍型时,才会丢弃包含的序列,这减少了由内含序列而引起的重叠群断点。第四,在初始图构建之后,hifiasm-meta使用测序深度信息来修剪unitig,假设来自同一菌株的unitigs往往具有相似的覆盖率。它还尝试连接来自不同单倍型的单元,以修补剩余的组装间隙。这些策略使hifiasm-meta在进行高精度宏基因组装时更加健全。
作者首先评估了hifiasm-meta (r58-31876a0),metaFlye (v.2.9) 和 HiCanu (v.2.2) 在两个人工合成菌群,ATCC和zymo(表 1),上的组装性能。ATCC由20个不同的物种组成,其中15个丰度较高,为0.18-18%,5个为稀有物种,丰度为0.02%。hifiasm-meta能够重建13个丰度较高的物种,并且每个物种都能被组装为一个完整的闭环重叠群,可与metaFlye和HiCanu相媲美。三种组装工具都将丰度为18%的牙龈卟啉单胞菌组装成两个重叠群,没有组装工具能够完全重建五个低丰度的物种。作者手动检查了这些低丰度物种的序列对齐文件,发现它们的组装间隙都是由于测序深度不足造成的,目前无法用现有的数据完全组装这些物种。zymo数据集包含17个不同种属的21个菌株,其中包括5个大肠杆菌,每个大肠杆菌的丰度约为8%。该数据集的挑战在于多个近缘物种大肠杆菌的同步出现。hifiasm-meta将菌株B3008组装成一个完整的闭环重叠群,菌株B766组装成两个重叠群,其余为碎片重叠群;HiCanu将B3008和B0组装成完整的闭环重叠群;metaFlye未能将五个菌株组装为闭环重叠群。hifiasm-meta将丰度为0.04%的硫代甲烷短杆菌组装为了更连续的重叠群。总体来说,三种组装软件在两个模拟菌群数据集上都具有相似的准确性。
表1. 评估组装的宏基因组数据集
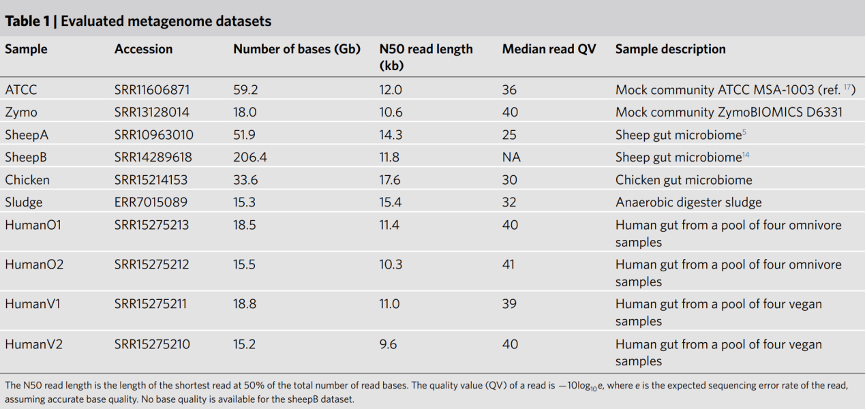
N50是基因组拼接之后一个评价指标,将拼接得到的所有的序列,根据序列大小从大到小进行排序,然后逐步开始累加,当加和长度超过总长一半时,加入的序列长度即为N50长度。
碱基的质量值(QV),在生物物理学中是碱基识别出错概率的整数映射,Q=-10*lgP,其中P为碱基识别出错的概率。
然后,作者在真实数据集上评估了三个HiFi宏基因组组装软件的性能(表1)。由于缺乏它们的真实成分信息,作者使用了CheckM评估每个组装数据集的完整度和污染水平。作者根据最低信息要求定义质量等级。从绵羊A肠道样本中,hifiasm-meta重建了323个长度超过1Mb的重叠群(图1a),其总长度为651 Mb。其中,根据CheckM的评估,176个MAGs定义为接近完整的基因组(图1b)。大多数未能达到接近完整的基因组的长重叠群是由于不完整,而不是由于污染。在176个接近完整的hifiasm-meta重叠群中,有134个是闭环的(图1b),与HiCanu(71个闭环的接近完整的重叠群)和metaFlye(47个)相比有明显的改进。我们将hifiasm-meta,HiCanu和metaFlye组装产生的重叠群相互对齐,研究了它们之间的相似性。作者发现分别有86%和94%的闭环的接近完整的HiCanu和metaFlye组装重叠群在hifiasm-meta组装中也是闭环的,并且长度相似。其余闭环的接近完整的HiCanu和metaFlye重叠群通过hifiasm-meta组装成了一个线性重叠群或两个线性重叠群。Hifiasm-meta可以重建其他组装软件产生的大多数高质量重叠群。此外,除了四对mash距离在0.62–0.92%之间的重叠群以外,其他hifiasm闭环重叠群之间的mash距离大多在1%以上。总体来说,高发散度(超过几个百分点)的菌株通常被组装为不相连的重叠群;少数低发散度的菌株用马赛克重叠群表示;许多混合发散的菌株可能导致复杂的装配图,并且组装起来最具挑战性。
为了从非环形重叠群中重建MAG,作者应用了MetaBAT2分箱算法到每个组装数据集。由于MetaBAT2未针对长读长组装数据进行优化,可能会错误地将同一物种的不同菌株分组到一个MAG中,甚至偶尔将两个闭环的重叠群分为一组,这些MAGs将被CheckM视为受到污染。为了改善分箱质量,作者将闭环的重叠群分成了单独的分箱基因组。最后,作者用三种组装软件从每个绵羊A的组装基因组中鉴定出了超过110个中等或高质量的非闭环MAGs(图1b)。而hifiasm-meta总是能鉴定更多的高质量MAGs。
随后,作者将hifiasm-meta应用于更大的绵阳B数据集中(表1),并获得了379个近乎完整的MAGs和279个闭环的重叠群。Bickhart等研究人员使用metaFlye组装了组合的绵羊A和绵羊B数据集,并结合Hi-C数据将组装的重叠群分箱为MAGs。他们报告了通过DAS Tool评估的44个闭环的重叠群和428个接近完整的MAGs。为了进行直接比较,作者在他们的组装结果上运行了 CheckM,并确定了241个接近完整的MAGs。比较下来,hifiasm-meta仅用HiFi数据便产生了一个更连续的组装。
对于鸡和四个人类肠道宏基因组数据集(表1),hifiasm-meta始终比HiCanu和metaFlye产生更多的闭环重叠群和更多的MAGs(图1b)。hifiasm-meta和metaFlye在污泥数据集上具有相当的性能,均优于HiCanu。相比较于绵羊A肠道样本,所有组装工具产生的MAGs都更少。为了了解这在多大程度上是由sheepA数据集的高数据量引起的,作者随机抽样了绵羊B,它代表与绵阳A相同的样本,但是在SequelII中测序,并且具有与humanO1相似的读长长度分布,其大约有18Gb的序列,与humanO1和污泥的数据集大小相当。在缩减的样本数据集上,作者可以组装出70个闭环的重叠群,远远超过humanO1和污泥中的闭环重叠群数量。这表明数据量确实会影响组装质量,但更连续的绵羊A装配可能与样本的组成更相关。
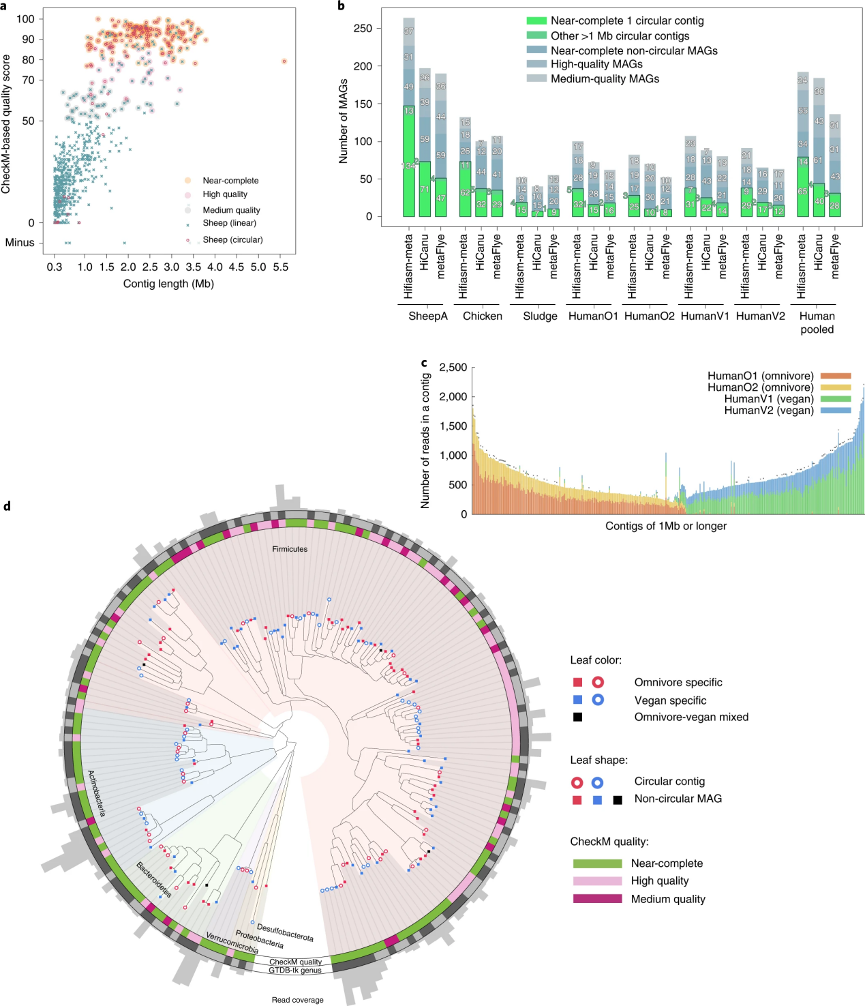
图1. 宏基因组组装实例数据。
a. 来自绵羊A样本的hifiasm-meta组装的长度超过300 kb的重叠群的质量得分。根据CheckM报告,污染度小于5%的MAG被保留。
b. CheckM评估结果。根据CheckM评估结果,完整性≥90%,污染水平≤5%的为“接近完整的MAG”;完整度≥70%,污染≤10%的则为“高质量MAG”;完整度≥50%的为“中等质量的MAG”。“HumanPooled”代表了四个人类肠道样本的共同组装。
c. 四个人类肠道样本中hifiasm-meta共同组装的长的重叠群的组成。每个柱子代表一个长度≥1 Mb的重叠群。每种颜色对应每个人类肠道样本。柱子顶部的标记表示重叠群是闭环的。
d. 来自共同组装的人类肠道MAGs的系统发育树。彩色分支对应于GTDB-Tk注释的不同门的细菌 。如果MAG中 90% 的序列分别来自杂食/素食志愿者样本,则MAGs是杂食/素食志愿者特异的。
在四个人类肠道数据集中,两个是从杂食志愿者那里收集的,另外两个是从素食主义者那里收集的。每个数据集代表一个由四个独立个体组成的混合数据(表1)。作者进一步将四个数据集汇集在一起并共同组装它们,通过在最终的 hifiasm-meta组装结果中报告的contig名称,作者可以根据序列来源去识别每个重叠群的组成。作者发现,绝大多数大≥1 Mb的重叠群,以及几乎所有≥1 Mb的闭环重叠群,要么是杂食志愿者特异性的,要么是素食主义者特有的(图1c),而两个杂食志愿者样品的重叠群充分混合了并不可区分,两个素食样品也是如此。
杂食志愿者和素食主义者的样本在共同组装的MAGs中也可很好地区分,尽管杂食志愿者和素食主义者的特异性MAGs在系统发育树中交叉存在(图1d);在这个系统发育树中,有20个属由三个或更多MAGs组成,其中17个属包含杂食志愿者和素食主义者特定的MAG。这表明hifiasm-meta组装更善于解开微生物之间微妙的组成差异。此外,值得注意的是,七个闭环重叠群的分支(在图1d的东北方向)经CheckM检验有75-79%的完整性,但它们都有5S,16S和23S核糖体RNA基因和大于18个转移RNA基因。
在计算性能方面,hifiasm-meta 在 18 个 CPU 线程上花费了 大约48 小时来组装sheepA和鸡数据集,而人类肠道样本则花费了大约3 小时。在这些数据集上,它与metaFlye一样快,并且始终比HiCanu快几倍。hifiasm-meta比 metaFlye 和HiCanu使用更多的内存,在进行sheepA和鸡肠样本组装时消耗大约200 Gb的内存。hifiasm-meta在8.9天内组装了最大的sheepB数据集,并在峰值使用了724 Gb内存。
在短读长测序时代,宏基因组组装很少被认为是重建全基因组的方法。但是这种观点因长读长组装的最新发展而改变。针对长而准确的HiFi读长进行了组装优化,hifiasm-meta进一步推动了宏基因组组装的发展。它可以在无需人工干预的条件下从一个深度测序的样品中组装更多的闭环MAGs,多于之前发布的所有闭环MAGs。这种高质量的宏基因组组装可能会从根本上改变宏基因组分析的实践,并揭示微生物群落的生物学和生物医学意义。
- 方 法 -
① hifiasm-meta算法概述
hifiasm-meta的工作流程包括可自定义的读长输入、序列纠错、序列重叠信息查找、字符串图构建和组装图清理。纠错和序列重叠步骤与原始 hifiasm 基本相同。我们添加了可选的读长选择,并改进了其余步骤。
② 输入读长的可选下采样
如果启用了读长选择,hifiasm-meta首先粗略地猜测对于整个数据集是否有太多的对齐计算要执行。这是通过检查锚点来完成的,并且不需要执行比对。如果三分之二的序列具有超过300个目标对齐序列,我们将继续进行选择。我们从一个空哈希表开始记录 k-mer 计数,并每记录2000个值进行一次遍历。在批处理中,对于遇到的每个读长,我们收集其规范的 k-mers 并查询哈希表以了解它们的出现情况。使用三个百分数,3%、5%和10%分别对照相应的阈值10、50和50进行检查。如果任何百分位数低于给定阈值,则保留序列。基本原理是,当有一些罕见的k-mers时,我们希望保留序列,因为当丢弃这些低频率的序列会导致信息丢失时。值得注意的是,这里的“稀有 k-mers”不一定全局罕见,如果输入被打乱,读长选择结果可能会发生变化。我们假设输入不是特别排序的。处理完批处理中的所有读长后,作者用它们更新k-mer计数哈希表(丢弃读长的 k-mers 也被计算在内)。读长选择的终止标准是保留的读长总数已超过所需计数,或者已处理所有序列。
③ 改进嵌合体检测
在组装图构建之前,原始的hifiasm将读长视为嵌合体,如果读长的中间部分未被其他读长覆盖,则丢弃它。由于统计波动,覆盖到低丰度基因组的读长可能具有这种未覆盖区域。hifiasm-meta的设定则是如果读长的两端与五个或更少的其他读长重叠,则禁用启发式搜索。这个额外的阈值提高了低丰度基因组的连续性。
④ contained reads的处理
构造字符串图的标准过程会丢弃较长序列中包含的更长的序列。如果contained reads和较长的序列实际上映射在不同的单倍型上,这可能会导致组装间隙。原始的hifiasm通过在图构建后通过contained reads来修补这些间隙,而hifiasm-meta是在图构建之前解决这个问题。如果推断出与序列完全重叠的其他序列来自不同的单倍型,则它保留contained reads。换句话说,hifiasm-meta只有在周围没有其他类似的单倍型时才会丢弃contained reads。此策略通常会保留实际上是冗余的额外的contained reads。这些额外的序列通常会导致bubble-like的子图,然后被原始hifiasm中的泡沫算法删除。
⑤ 宏基因组数据集的组装
作者评估了使用了48个CPU线程评估了hifiasm-meta r58,HiCanu v.2.2和metaFlye v.2.9。使用“hifiasm-meta read.fa”参数来组装经验肠道样本,使用“hifiasm-meta --force-rs read.fa”参数来为两个模拟菌群数据集启用读长选择,用“canu maxInputCoverage=1000 genomeSize=100m batMemory=200 -pacbio-hifi read.fa”参数运行HiCanu。作者将绵羊A的“基因组大小”参数增加到了1000m,并得到了相同的结果。作者用“flye --pacbio-hifi read.fa --plasmid --meta”参数运行了metaFlye。记录hifiasm-meta和metaFlye组装时间和峰值内存使用量。同时,作者使用脚本(https://gist.github.com/xfengnefx/d4abf19de8ebae9cc8ccd56e9898604d)来检查/proc/ID/status文件来衡量HiCanu的性能。对于其他的一般文件操作,作者使用了seqtk(https://github.com/lh3/seqtk,1.3-r107-dirty),readfq.py(https://github.com/lh3/readfq,7c04ce7),GNU Parallel和SAMtools等软件。
⑥ 宏基因组分箱
作者使用了MetaBAT2进行初始分箱,然后对MetaBAT2结果进行后续处理以获得最终的MAG。作者使用“minimap2 -ak19 -w10 -I10G -g5k -r2k --lj-min-ratio 0.5 -A2 -B5 -O5,56 -E4,1 -z400,50 contigs.fa read.fa”方法将原始数据对齐到了组装数据集种,并使用“jgi_summa_rsize_bam_contig_depths --outputDepth depth.txt input.bam”计算了组装基因组的测序深度,同时,使用“metabat2 --seed 2 -i contigs.fa -a depth.txt”运行 MetaBAT1。作者也尝试了不同的随机种子或“-s 500000”参数,并得到了类似的结果。作者仅将MetaBAT2应用于主要的hifiasm-meta和HiCanu组件。在 MetaBAT2 分箱后,如果将 1 Mb 或更长的闭环重叠群与其他重叠群分箱在一起,作者将拆分它为一个单独的MAG。
⑦ 评估模拟宏基因组文库的组装结果
为了评估组装的质量,作者将带有“minimap2 -cxasm20”的重叠群映射到参考基因组,并检查了比对的结构变化。在从两个模拟群落组装的22个闭环hifiasm-meta重叠群中,除了一个链球菌变种外,有21个与参考一致。对于这个基因组,hifiasm-meta引入了一个20 kb的缺失,该缺失得到了一小部分对齐的读长的支持,这表明这是菌群中真实但罕见的等位基因。
在重叠群与参考序列的比对中,作者观察到每个基因组有数千个碱基的错配和缺失。而HiFi的组装结果之间的差异数量要少得多。例如,对于脑膜炎奈瑟菌,hifiasm-meta重叠群和参考基因组之间存在6090个小差异,但hifiasm-meta和HiCanu重叠群之间只有两个小的碱基对差异。我们怀疑这6090个差异中的大多数可能是参考基因组中的常见错误。
⑧ 评估宏基因组组装
作者运行了CheckM v.1.1.3来评估MAGs的完整性和污染水平。命令行是 “checkm lineage_wf -x fa input/ wd/;Checkm qa -o 2 wd/lineage.ms”。作者还尝试了DAS Tool用于对 sheepA 数据集进行评估。与CheckM相比,DAS Tool的结果更加乐观,识别出的接近完成的MAGs比CheckM多 22%。由于CheckM更常用于评估,因此作者仅将CheckM应用于所有的组装数据集。此外,对于绵羊B数据,yak QV用于评估重叠群的正确性。
作者使用GTDB-Tk v.1.3.0及其数据库版本r95通过命令“gtdbtk classify_wf”进行系统发育树构建。作者注释了树并使用GraPhlAn进行了可视化。
作者使用了INFERNAL从重叠群中鉴定rRNA和tRNA。命令行是“cmscan –cut_ga –rfam –nohmmonly –fmt 2 –tblout cmscan.table Rfam.cm in.fa”。E 值大于 0.01的条目被删除。在本文中,由hifiasm-meta组装的733个长的闭环重叠群中共有738个具有完整的RNA,即所有三种类型的rRNA至少有一个全长拷贝,以及至少18个tRNA具有完整长度。
报告摘要
有关研究设计的更多信息,请参阅本文链接的《自然研究报告摘要》。
数据公开
表1展示了来自NCBI原始数据数据库(SRA)的HiFi数据。所有生成的组装结果和图形的基础数据都可以在https://zenodo.org/record/6330282 上找到。
ZymoBIOMICS模拟参考基因组是从https://s3.amazonaws.com/zymo-files/BioPool/D6331.refseq.zip下载的。
ATCC模拟菌群中的参考基因组可在https://www.atcc.org/products/msa-1003 获得。
CheckM数据库:
https://data.ace.uq.edu.au/public/CheckM_databases/checkm_data_2015_01_16.tar.gz。
GTDB-Tk 数据库:
https://data.ace.uq.edu.au/public/gtdb/data/releases/release95/95.0/auxillary_files/。
代码公开
Hifiasm-meta的源代码可在 https://github.com/xfengnefx/hifiasm-meta 获得。
参考文献
Feng, X., Cheng, H., Portik, D. et al. Metagenome assembly of high-fidelity long reads with hifiasm-meta. Nat Methods 19, 671–674 (2022). https://doi.org/10.1038/s41592-022-01478-3
- 第一作者简介 -

哈佛大学医学院Dana-Farber癌症研究所
Xiaowen Feng
博后
第一作者:Xiaowen Feng,哈佛大学医学院Dana-Farber癌症研究所博后,主要做三代组装算法的开发。目前以第一作者发表了1篇Nature Methods,1篇Molecular Biology and Evolution。
- 通讯作者简介 -

哈佛大学医学院Dana-Farber癌症研究所
李恒
副教授
通讯作者:李恒,哈佛大学医学院,Dana-Farber癌症研究所副教授。于南京大学获得物理学学士学位,并于2006年在中国科学院理论物理研究所获得理论生物物理学博士学位。曾在威康桑格研究所(Wellcome Trust Sanger Institute)做博士后,2009年开始在博德研究所(Broad Institute)工作。于2018年加入Dana-Faber癌症研究所和哈佛医学院。主要研究方向为利用先进的计算方法来分析大规模的生物序列数据,并解决生物医学研究中的实际问题。个人主页:(https://www.dana-farber.org/find-a-doctor/heng-li/)
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读