目录
- 0. 导论
- 基本概念
- 1. 生物标志物发现的注意事项
- 2. 数据预处理
- 2.1 高质量原始数据和缺失值处理
- 2.2 数据过滤
- 2.3 数据归一化
- 3. 数据质量评估
- 3.1 混杂因素
- 3.2 类别分离
- 3.3 功效分析
- 3.4 批次效应
- 4. 生物标志物发现
- 4.1 策略
- 4.2 数据分析工具
- 4.3 模型优化策略
0. 导论
组学技术有望改善精准医学中生物标志物的发现。已发现的生物标志物的首要问题是在不同队列之间的无法重现。从数据分析角度来看,主要原因是统计方法的偏差,以及批次效应和混杂因素导致的过拟合。可重现生物标记物发现的关键在于:恰当的实验设计、无偏的数据预处理和质量控制分析,以及对统计学和机器学习算法的熟练应用。
该篇综述讨论了实验设计和分析的注意事项,并从专家角度推荐了一些标准。
基本概念
精准医学(precision medicine):一种根据基因、环境和生活方式的个体差异来调整疾病治疗和预防策略的举措。这些差异会产生不同的疾病表型,即疾病的异质性。
生物标志物(biomarkers):能够区分不同人群(如健康人和患者,肿瘤良性和恶性,是否响应特定疗法)、可测量的生物分子(即基因、蛋白和代谢物)。
特征(signature):生物标志物在组合使用时最为有效,这种组合被称为特征。
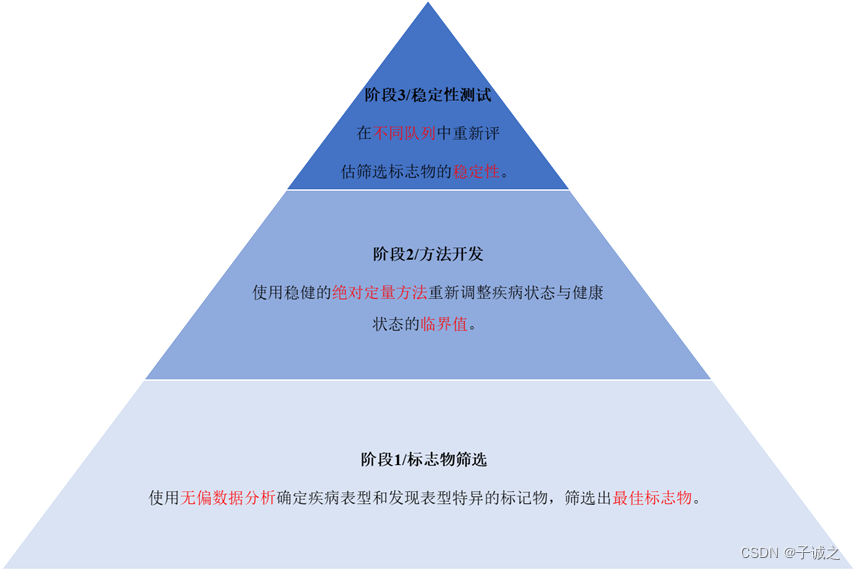
图1. 精准医学中生物标志发现的不同阶段。第一阶段,利用无偏数据分析探索健康大数据,进行表型特异的生物标志物发现,筛出最佳分析物。第二阶段,使用稳健的绝对定量方法重新调整疾病与健康的cut-off值。第三阶段,在不同的队列和更多的人群中重新评估标志物的稳定性。
临床研究类型[2]:据不同的研究目的,临床研究有不同的类型,分别用于产生假设,检验假设,验证假设。
-
观察性研究:研究过程中未对受试者施加人为干预措施。
- 描述性研究:仅描述一个事件或现状,不能作为病因分析的直接证据,目的是产生假设,为进一步分析性研究提供参考。没有对照组,论证强度较弱,可重复性差。
- 病例报告和病例系列:对一两个或多个的病例进行记录和描述,试图在疾病的表现、机理以及诊断治疗等方面提供医学报告。
- 横断面研究:研究特定时期,暴露因素和疾病或者健康状况的关系。研究结果仅为因果联系提供线索。
- 分析性研究:分析性研究有对照组。包括病例对照研究(结局→暴露因素)、队列研究(暴露因素→结局)以及横断面研究。用以产生假设和检验假设。
- 病例对照研究:选定病例组(患者)和对照组(正常人群或未患该病的人群),分别研究两组在过去暴露于某个危险因素的情况,进而判断暴露的危险因素与疾病的相关性及强度。是一种回顾性研究。用于探索病因,筛选危险因素和检验假设。
- 队列研究(狭义?):将人群按照暴露于或未暴露于某种因素(包括危险因素、致病因素等)分为两组,之后经过追踪一段时间后比较两组发病或死亡的结局,从而判断暴露的危险因素与疾病的相关性及强度。是一种前瞻性研究。用于检验病因假设。
- 描述性研究:仅描述一个事件或现状,不能作为病因分析的直接证据,目的是产生假设,为进一步分析性研究提供参考。没有对照组,论证强度较弱,可重复性差。
-
实验性研究:研究过程中对受试者施加一定的人为干预措施,从而观察干预因素对于研究对象结局的影响。也是一种前瞻性研究。三大要素:对象、干预、结局。五大原则:对照、重复、随机、均衡、盲法。
- 随机对照研究:采用随机的方法,将合格的研究对象随机分到试验组和对照组,继而分别接受相应的干预措施,观察干预措施的效应,并采取客观的指标衡量试验结果。最常应用于干预性研究和药物评价。
- 非随机对照研究:非随机分组。
另外,前瞻性队列研究与回顾性队列研究的区别在于作为观察终点的事件在研究开始时是否已发生。
1. 生物标志物发现的注意事项
实验设计
实验设计在消除数据集偏差方面起着至关重要的作用。
在人群研究中,基于临床变量(如年龄、性别、种族)进行配对被认为是一种非常有效的策略,可以最大限度地减少许多混杂因素的影响。然而,过度配对可能会导致强标志物的丢失,并降低生物标志物的区分力。例如,BMI是肥胖相关代谢紊乱的一个强决定因素。因此,在与肥胖引起的代谢性疾病相关的研究中,不建议通过配对策略来抵消BMI的区分力。
队列选择策略因研究类型(前瞻、回顾、横断面)而异。无论何种类型研究,都需要在选择高风险队列和无风险队列之间取得平衡。使用具有高患病风险的特定人群来发现生物标志物,会降低对普通人群的代表性。在发现预测性标志物时,在疾病的早期阶段设计很重要,此时病例和对照之间的分级不明显或紧密重叠。选择无风险队列也不能很好地代表普通人群。统计学上最好的方法是设计一个可以反映整个人群或特定亚人群的实际疾病比例的研究。例如,如果一种疾病在整个人群中的发病率为10%,那么理想的配对病例对照研究应为每个病例匹配9个对照组。然而,由于病人的可及性,这种研究设计可能并不实用。因此,研究设计应尽可能反映实际的疾病比例。
生物样本质量
生物样本的质量受采集过程、储存条件、运输和重复使用的影响,这些因素都会极大地影响生物标记物发现的重现性。生物样本采集应由经过培训的人员按照SOP进行。生物样本应该用专门的试管收集。然后必须将这些试管储存在设计良好的生物库或生物储藏室中。生物库的推荐温度在-20℃ ~ -70℃。在生物样本的运输过程中,温度控制也是必要的。在试管贴上适当的标签对于检索参与者信息非常重要。样本反复冻融会导致某些分析物降解,这会导致固有批次效应。为了解冻融循环导致的降解趋势,可通过多次冻融循环,使用多次时间采样进行试点研究,以确定冻融循环中可能出现的分析物,并可用于批次效应校正。
2. 数据预处理
2.1 高质量原始数据和缺失值处理
高质量原始数据*虽然在技术上是最稳健的,但通常有大量缺失值。缺失值通常是由于样本的浓度、信号低于检测限,或信息缺失造成的。处理缺失值主要有两种方法:删失或填充。最佳方案取决于研究目的、对数据和分析方法局限性的正确理解、以及给定变量的缺失值百分比(通常数据缺失 >10-15% 的变量被认为是可以删除的,但也要考虑数据集的大小)。
在定量生物标志物发现研究中,如果缺失值是由于浓度或信号低于阈值造成的,最好是删除缺失值的变量。在定性生物标志物发现研究中,可以对这些缺失值进行估计。对于低于检测限的估计值,标准做法是用检测到的最小阳性值的一半来替代。对于其他情况,也可以用变量均值、KNN或回归法替代。
* Stringent data直译为严格数据,即选择最高置信度分数生成的原始数据。比如测序中的高质量碱基,为了更加直观,这里意译为高质量数据。
2.2 数据过滤
数据过滤用于识别和删除噪声以及无意义变量,从而提高发现类间真实差异的分析功效。数据过滤可分为无模型技术(如均值、四分位数区间、标准差等)和基于模型的技术(如卡尔曼滤波、粒子滤波等)。
在发现用于预测的标记物研究中,不建议从小数据集中过滤掉无意义数据,直到数据不干扰最小阈值为止。对于可变噪声通常很高的大数据集,数据过滤是一个值得考虑的选项。有必要过滤仪器产生的噪声,例如,非靶向NMR数据集通常有许多接近基线的峰,这些峰可能是技术噪音,可以通过均值或中位数识别。标准差或四分位间范围(IQR)可识别稳态变量*,这些变量应从数据集中排除。在分析设备(LC-MS、GC-MS等)输出的数据中,相对标准差RSD(SD/mean)通过参考样本计算得出,RSD百分比高的变量被视为噪声。
* 稳态变量(homeostasis variables)是指在整个研究过程中数值几乎恒定的变量。
2.3 数据归一化
数据归一化的策略包括应用规则、数据转换和数据缩放。归一化数据集如果没有明显的数据冗余,且缩放合理,通常具有钟形分布。可以使用简单的数据分布分析(或正态检验)来衡量数据的正态性。归一化数据或正态数据更适合线性方法和标准的统计方法。如果无法实现数据正态分布,可以使用非参数统计方法。
用于数据归一化的规则大致分为局部和全局两类。局部归一化基于参考变量*进行。使用变量作为参照会产生内在偏差。当在多个变量中选择时,对变量进行简单线性回归可以量化它们各自的分类能力。分类能力最弱的变量被认为是最稳定的,可用于归一化。除了单个参考,也可以使用一组参考的求和、均值或中位数计算参考池。全局归一化不依赖于任何特定的参考。它计算所有样本,并根据特定规则创建自己的参照。Z-score是一种常见的全局归一化技术,数据经过对数转换和缩放,数据分布的均值为0,标准差为1。Q-norm是另一种常见的多样本归一化技术,它将观测到的样本间变异归为技术原因而非生物学原因。Q-norm计算各样本中每个分位数的均值,然后将每个分位数均值作为参考,并将所有观察到的分布均等化为所有样本的一个平均分布。由于Q-norm没有考虑到生物条件导致的样本间观测差异,因此会降低功效。为了解决这一缺陷,Hicks等提出了平滑分位数归一化,即假定每个样本的统计分布在给定的生物条件下,而非所有样本中,是相同的。
如果数据无法通过应用规则成功归一化,需要进行数学转换。常用的数据转换包括对数转换、立方根变换、反双曲正弦(arcsinh,能处理零值和负值)。
如果数据转换无法建立正态分布,则有必要进一步应用规则或采用数据缩放。缩放技术包括:以均值为中心的缩放;以均值为中心除以每个变量的标准差或标准误;以均值为中心除以每个变量的范围。
* 参考变量被认为是标准数据,所有其他数据都要与之对齐。参考变量应在疾病过程中保持稳定。
3. 数据质量评估
在数据分析前,实验设计需要从混杂因素、功效和批次效应方面进行优化。化学计量分析被广泛应用于数据质量的评估和调整。
3.1 混杂因素
由于实验设计时无法考虑到每一个影响因素,导致人群健康数据中无可避免地存在一些混杂因素。混杂因素影响较大时,会导致结果或解释出现偏差。虽然无法完全消除,但有效的策略可以将这种不必要的影响降至最低。这些策略主要是高维数据的可视化方法。它们可以从具有不同依赖性和关系的变量集合中创建了一组独立变量(即成分),成分通过消除冗余,以容易理解的方式展示高维数据。其中一些方法还能识别混杂因素是否存在及其加权影响。
许多统计可视化工具可以识别数据集中混杂因素的影响。其中,主成分分析(PCA)是最常见的方法。PCA以无监督的方式对多维数据进行正交聚类,以确定主成分/特征向量与其大小/特征值之间的线性关系。数据中贡献百分比最高的主要PC通常代表已知的生物条件,如病例和对照。每个非主要PC的贡献百分比通常较小。任何贡献率>10%的非主要PC都被视为主要混杂因素,会对最终数据解释产生重大影响,并导致结果偏差。如果存在此类混杂因素,建议重新设计研究(如增加更多配对标准)。除了描述混杂因素的影响外,PCA还可用于从数据集中选择性地排除已知的与混杂因素相关的聚类,也被称为降维。通常去除特征值<=2的PC,但在生物标记物发现研究中并不推荐这种降维方法,因为可能会导致结果偏差。因此,对于特征值较小的非主要PC,无需通过降维将其从数据集中去除。
t-SNE是另一种常见的可视化方法,它有时会被用于复杂的数据集,作为一种探索性选择,以显示PCA无法观察到的非线性关系。由于t-SNE使用的是概率公式,具有一定的随机性,因而是不可再现的。
3.2 类别分离
主要PC的累积贡献百分比(特征值)应代表疾病模型数据集中的大部分方差,而这在预测模型数据集中可能并不突出,这取决于研究疾病的早期程度。如果主要PC的累积贡献率过低,下游分析就很有可能失败。因此,有必要使用经验贝叶斯方法了解数据集中病例与对照之间的类别分离强度。首先,使用监督统计聚类方法(PLS-DA、sPLS-DA、orthoPLS-DA)估计两个实验组别的分离度。之后,使用经验贝叶斯方法的置换检验重新确认估计的类别分离度。用于计算随机置换检验中无类别分离概率的比例的经验p值至少应<0.5,以确保类别分离不是随机事件。
3.3 功效分析
功效分析(power analysis)是一种用于理解研究在预测功效方面的人口代表性的统计方法,应在研究设计阶段进行。它决定了一项研究在给定置信度内检测效应值的强度。换句话说,这种分析可以预测在一定置信度内取得给定效应值所需的样本量。鉴于数据集的功效主要受效应值和样本量影响,一般来说,高效应值和大样本量会提高研究的功效。虽然统计功效为0.8被认为是有效研究的基准,但如果研究功效处于曲线的指数阶段,作为试点研究,低功效也是可接受的。这一阶段代表了非常高的效应值,增加几倍的样本量就足以达到0.8的研究功效。
3.4 批次效应
批次效应是指由于实验室条件、平台、试剂批次和人员变化而产生的实验失真,导致各组样本或批次间出现系统的、非生物学的差异。在大多数研究中,批次效应都会被忽略。然而,这种现象往往会影响数据的整合与解读,并可能将结果转化为错误的发现。因此,在没有进行批次效应分析的情况下,将两个或多个批次的数据合并是不恰当的。
为了处理批次效应问题,标准方法是在每批次的每个组别中跑一些相同的样本,即参考样本。不同批次间参考样本的线性比较可用于观察和估计批次效应造成的差异。如果在不同批次间观察到批次效应,则必须对其进行相应调整。虽然上述方法可以了解批次效应,但不建议通过线性模型的估算校正批次。ComBat是对不同类型的生物数据集进行批次效应可视化和校正的最常用方法。该算法使用参考样本,通过一个批次中给定变量与所有批次中该变量平均值的距离来计算方差。然后,通过经验贝叶斯算法调整尺度以校正方差。
在没有参考样本的情况下,不需要变异来源是未知的,SVA可用于估算这种未知的变化。在SVA中,参考样本的替代物是根据不同批次中不常见的差异表达变量确定的。从生物学角度来看,理论上不同批次的病例和对照之间的差异表达变量应该是一致的。然而,这些不常见变量(替代变量,surrogate variables)的存在可能是由批次效应引起的。分析替代变量可以估计批次效应,然后用ComBat对批次效应进行校正。
4. 生物标志物发现
4.1 策略
从策略上讲,第一阶段的生物标志物发现分为两类:定性和定量。定性方法采用变量排序法,而定量方法则根据浓度临界值选择变量。
定性方法包括支持向量机(SVM)、多元逻辑回归(MLR)、PLS-DA、随机森林等。PLS-DA中变量的排序基于其VIP分数,VIP>1可认为重要,但在选择变量或标志物时,VIP>2更为可取。MLR中变量的排序基于其AUC。从排名靠前的变量中,筛选出一些变量组合作为特征,以提高AUC。虽然这些定性方法不能为新受试者的决策提供即时的量化判断,但却是缩小生物标志物范围的最佳方法。
逻辑回归是一种常见的单变量定量生物标志物发现方法,其使用单变量对人群进行分类。决策树是一种常见的多变量定量生物标志物发现方法,其将多个生物标志物以树的形式聚类,并带有量值和方向。
4.2 数据分析工具
(1)统计工具
简单的回归分析(如逻辑回归、线性回归等)可以衡量单一变量的类别区分能力。虽然单一生物标志物更适用于临床环境,但这在很大程度上并不现实,尤其对糖尿病、心血管疾病、癌症等慢性疾病而言。相比之下,多变量分析更适合在发现生物标志物特征时选择多个变量。
逐步多元逻辑回归(sMLR)是多变量ROC分析中一种经典的统计工具。它通过对每个变量进行逻辑回归排出最佳预测因子,然后将这些变量组合起来,以达到最大的预测性。有时,数据集的解释变量间可能存在线性依赖关系,引发多重共线性问题,导致sMLR筛出更多的冗余变量。因此,应通过置换分析仔细检查sMLR筛出特征的冗余程度。前面提到的PLS-DA是多元ROC分析中特征/变量排序的另一种统计工具。虽然它在降维分析中容易对数据过拟合,但它可以通过适当的自举法(详见模型优化)来处理排序过程中的数据冗余问题。
(2)机器学习
机器学习方法是另一种常见的多元ROC分析方法集。它使用不同的算法从一个数据子集(训练集)中学习,然后在另一个数据子集(测试集)上验证其预测模型。样本量和自变量-因变量关系是选择机器学习算法时需要考虑的两个重要因素,线性算法通常适合小样本量、线性关系的数据集,而非线性算法则能够使用大样本量,对非线性关系进行分类。例如,SVM是一种有监督的线性机器学习算法,它可以通过寻找能区分两个类别的超平面,对相对较小的数据集进行分类。
贝叶斯网络(BNet)可用于复杂疾病的中大型数据集。BNet可根据结果概率计算原因概率,其表示联合概率分布的有向无环图可以衡量给定类别的概率。遗传算法(GA)是另一种非线性算法,尤其适用于因内部过程复杂而导致结果高度不可预测的情况。其它常见的非线性机器学习算法还有基于决策树的算法,如随机森林等;集成算法,如投票、堆叠等;深度学习等。一般来说,非线性算法对样本量的要求比较灵活,纳入更多数据可以提高其准确分类的能力。不过每种方法的准确率都有一定的上限,之后就会趋于稳定。样本量范围取决于数据类型。这些非参数算法可以自行确定建立分类模型所需的参数。
值得注意的是,许多算法只适用于特定类型的数据。可以对数据类型进行调整,以兼容所选算法。此外,在同一数据集上探索不同的算法非常重要,进而找出最合适的算法,从而建立在AUC、准确率、灵敏度和特异性方面最可靠的模型。不过,这些模型通常还需要进一步优化。
4.3 模型优化策略
每个模型都可能存在过拟合和选择偏差的问题。为减少此类问题的发生,建议采用一系列验证过程和参数调整。验证的选择取决于数据集的大小。对于大型数据集(>200个受试者),可将受试者分为训练集(60%)和独立测试集(40%)。对于小型数据集(<200个受试者),如果创建测试数据集,则获得优化模型的几率较低。建议将所有可用数据纳入训练集,并执行严格的验证方案。
对于使用小数据集的统计模型,如逻辑回归、sMLR、PLS-DA等,自举法为标志物和特征发现提供了严格验证。自举法采用有放回的随机抽样生成模型,最终结果由模型的平均值计算得出。对于使用小数据集的机器学习方法,K折交叉验证(K-fold CV)是验证的首选方法。通常情况下,较高K值会导致模型过拟合,而较低的k值则会导致拟合效果不佳或产生高度可变和有偏差的模型。K值选择根据"一个标准误规则"进行优化。即优化后K模型的误差应较低(误差小),且不应大于准确度饱和点(最简单)。
对于大型数据集,可使用独立测试集进一步检验训练集中的优化模型。模型优化策略如下图所示。原则上,在同一研究队列训练集中的优化模型在独立测试集中的表现应该相似。如果两者差异很大,则表明训练集中的模型优化不当。
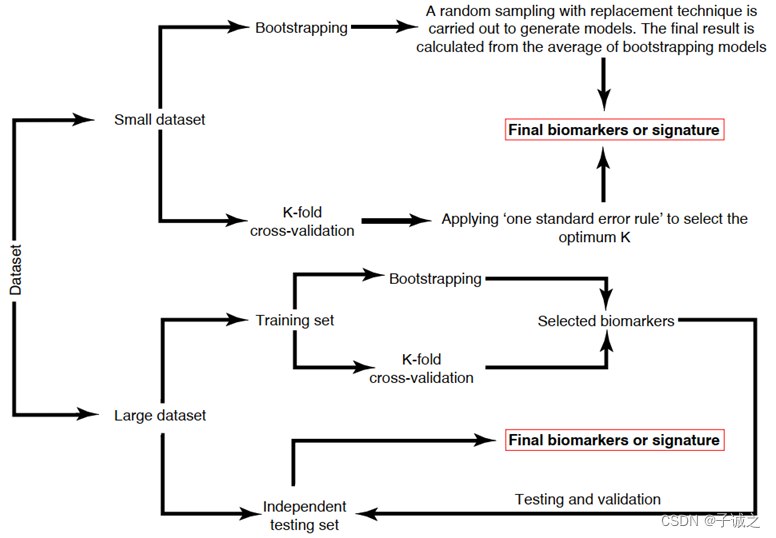
图2. 基于数据集规模的模型优化策略。在大型数据集中,数据被分为训练集(60-80%)和独立测试集。在测试集中,采用适当的验证方案(如K折交叉验证和自举法)来选择生物标志物。在训练集中发现的生物标志物将在独立测试集中进一步验证。在小型数据集中,一个独立测试集不可行。因此,需要通过 K 折交叉验证或自举法来考虑生物标志物的可靠性。
参考文献
[1] Khan, Saifur R., et al. “Unbiased data analytic strategies to improve biomarker discovery in precision medicine.” Drug Discovery Today 24.9 (2019): 1735-1748.
[2] 临床研究的类型:http://www.mengyin.gov.cn/info/8309/128766.htm
时间线
20231010:主要翻译自[1],有少量删改。
20231014:新增临床研究概念[2]。





![VCAP-DCV VMware vSphere: 运维、扩展和安全防护 [V8.0]](https://img-blog.csdnimg.cn/a8fb31ac625345e5a38b7e5b5a73693b.png)