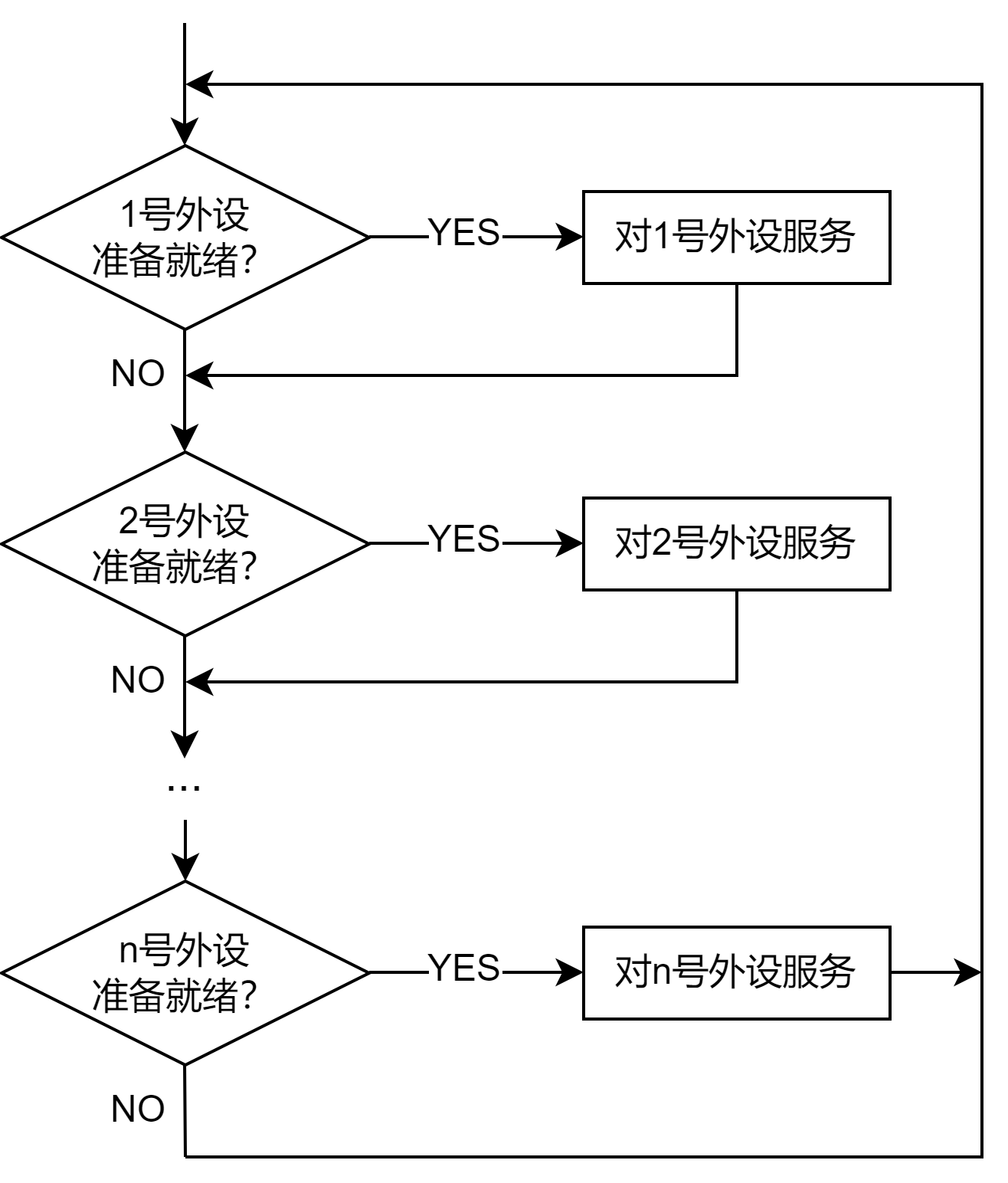
早在1997 年出台的21 CFR Part 11中,就对与药物产品审批放行相关的电子记录有规定,应采取与纸质记录相同的控制要求1,包括生成或维护电子记录须采用恰当的访问控制措施,记录变更必须体现在记录审计追踪中等。
2003年,FDA发布了一项指导文件强调 21 CFR Part 11中规定的要求应主要针对实现批次记录所需的相关记录以及可影响产品放行决策的相关记录2。

EU GMP Annex 113针对访问与签名控制措施的核心要素中加入了风险评估和系统设计或技术参数,从而将电子数据和电子签名要求的应用范围进行了扩展。
2018 年,FDA向业界发布了一份指导文件“数据完整性与药品cGMP合规性问答6”,根据21 CFR Part 210、211 和 212要求,明确了数据完整性在现行良好生产规范 (cGMP)中的作用。该指导文件对访问控制、规定的静态和动态记录以及其他符合相关规则的操作要求进行了更详细的定义。FDA要求所有数据均准确可靠,而cGMP指南要求根据风险采取相关策略,以便检测和预防数据完整性问题。
与此同时,欧盟重新审查了电子记录、电子签名和英国MHRA4的指导方针,均基于ALCOA+原则。
2020年12月,中国国家药监局(NMPA) 开始施行药品记录与数据管理要求(试行)规定,其基本要求中指出,为保障药品质量和患者安全,必须在“从事药品研制、生产、经营、使用活动中,保证全过程信息真实、准确、完整和可追溯”,也与ALCOA+原则相一致。
过滤器的测试原理:
1、起泡点法测试原理:
当滤膜和滤芯用一定的溶液完全浸润,然后通过气源在一侧加压(我们仪器里面有进气控制系统,可以稳定压力,调节进气),随着压力的增加,气体从滤膜的一侧放出,表现膜一侧出现大小、数量不等的气泡,通过仪器判断出对应的压力值就是泡点;
2、扩散流法测试原理:
扩散流测试是指当气体压力在滤芯起泡点值的80%时,这时还没有出现大量的气体穿孔而过,只是少量的气体先溶解到液相的隔膜中,然后从该液相扩散到另一面的气相中,这部分气体称之为扩散流;
3、为什么扩散流的方法更好:
起泡点值只是一个定性的值,从开始起泡到最后的群起泡是一个比较长的过程,不能准确的定量,而测量扩散流值是一个定量值,不但能准确的确定过滤器的完整性,而且还能反应出膜的孔隙率、流量和有效过滤面积等方面的问题,这也就是为什么国外厂家都用扩散流法测试完整性的原因;
4、水侵入法测试原理:
水侵入法专用于疏水性滤芯的测试,疏水性膜抗拒水,孔径越小,把水挤入疏水膜中需要的压力越大,所以在一定的压力下,测量挤入滤膜中的水流量来判断滤芯的孔径。
悬浮液中的固体颗粒大、粒度均匀时,过滤的滤渣层孔隙较为畅通,滤液通过滤渣层的速度较大,应用凝聚剂将微细的颗粒集合成较大的团块,有利于提高过滤速度,对于固体颗粒沉降速度快的悬浮液,应用在过滤介质上部加料的过滤机,使过滤方向与重力方向一致,粗颗粒首先沉降,可减少过滤介质和滤渣层的堵塞;在难过滤的悬浮液(如胶体)中混入如硅藻土、膨胀珍珠岩等较粗的固体颗粒,可使滤渣层变得疏松;滤液粘度较大时,可加热悬浮液以降低粘度。这些措施都能加快过滤速度。
北京泽平科技代理各大品牌完整性检测技术产品,请搜索“北京泽平科技”进入咨询。






![[洛谷]P1449 后缀表达式](https://img-blog.csdnimg.cn/e2b30d39b81b46d4a5f7f27cb99079c3.png)