单细胞之富集分析-6:PROGENy - 简书 (jianshu.com)
#request 2
.libPaths(c( "/home/data/t040413/R/x86_64-pc-linux-gnu-library/4.2",
"/home/data/t040413/R/yll/usr/local/lib/R/site-library",
"/home/data/refdir/Rlib/", "/usr/local/lib/R/library"))
## We load the required packages
library(Seurat)
library(decoupleR)
# Only needed for data handling and plotting
library(dplyr)
library(tibble)
library(tidyr)
library(patchwork)
library(ggplot2)
library(pheatmap)
library(CellChat)
library(patchwork)
library(ggplot2)
library(ggalluvial)
library(svglite)
library(Seurat)
library(xlsx)
library(harmony)
#https://saezlab.github.io/progeny/#:~:text=PROGENy%20is%20resource%20that%20leverages%20a%20large%20compendium,infer%20pathway%20activities%20from%20bulk%20or%20single-cell%20transcriptomics.
#https://cloud.tencent.com/developer/article/2206142
#https://saezlab.github.io/decoupleR/articles/decoupleR.html
library(progeny)
library(Seurat)
#https://saezlab.github.io/decoupleR/articles/pw_sc.html
inputs_dir <- system.file("extdata", package = "decoupleR")
data <- readRDS(file.path(inputs_dir, "sc_data.rds"))
DimPlot(data, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
#BiocManager::install("OmnipathR")
library(decoupleR)
net <- get_progeny(organism = 'human', top = 500)
net
table(net$source)
# Extract the normalized log-transformed counts
mat <- as.matrix(data@assays$RNA@data)
# Run mlm
acts <- run_mlm(mat=mat, net=net, .source='source', .target='target',
.mor='weight', minsize = 5)
acts
# Extract mlm and store it in pathwaysmlm in data
data[['pathwaysmlm']] <- acts %>%
pivot_wider(id_cols = 'source', names_from = 'condition',
values_from = 'score') %>%
column_to_rownames('source') %>%
Seurat::CreateAssayObject(.)
# Change assay
DefaultAssay(object = data) <- "pathwaysmlm"
# Scale the data
data <- ScaleData(data)
data@assays$pathwaysmlm@data <- data@assays$pathwaysmlm@scale.data
p1 <- DimPlot(data, reduction = "umap", label = TRUE, pt.size = 0.5) +
NoLegend() + ggtitle('Cell types')
p2 <- (FeaturePlot(data, features = c("Trail")) &
scale_colour_gradient2(low = 'blue', mid = 'white', high = 'red')) +
ggtitle('Trail activity')
p1 | p2
table(net$source)
net[net$source=="Trail",]$target
rownames(data)
data=AddModuleScore(data,features = list(net[net$source=="Trail",]$target),assay = "RNA")
#单细胞progeny-----
#https://cloud.tencent.com/developer/article/2206142
1
data("pbmc3k")
pbmc3k.final <- pbmc3k
pbmc3k.final[['percent.mt']] <- PercentageFeatureSet(pbmc3k.final, pattern = '^MT-')
pbmc3k.final <- subset(x = pbmc3k.final, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc3k.final <- NormalizeData(pbmc3k.final)
pbmc3k.final <- FindVariableFeatures(pbmc3k.final)
pbmc3k.final <- ScaleData(pbmc3k.final, features = rownames(pbmc3k.final))
pbmc3k.final <- RunPCA(pbmc3k.final, features = VariableFeatures(pbmc3k.final))
pbmc3k.final <- JackStraw(pbmc3k.final)
pbmc3k.final <- ScoreJackStraw(pbmc3k.final, dims = 1:20)
pbmc3k.final <- FindNeighbors(pbmc3k.final, dims = 1:10)
pbmc3k.final <- FindClusters(pbmc3k.final, resolution = 0.5)
pbmc3k.final <- RunUMAP(pbmc3k.final, dims = 1:10)
new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc3k.final)
pbmc3k.final <- RenameIdents(pbmc3k.final, new.cluster.ids)
pbmc=pbmc3k.final
# We create a data frame with the specification of the cells that belong to
## each cluster to match with the Progeny scores.
CellsClusters <- data.frame(Cell = names(Idents(pbmc)),
CellType = as.character(Idents(pbmc)),
stringsAsFactors = FALSE)
head(CellsClusters)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
2
## We compute the Progeny activity scores and add them to our Seurat object
## as a new assay called Progeny.
pbmc <- progeny(pbmc, scale=FALSE, organism="Human", top=500, perm=1, return_assay = TRUE)
pbmc@assays$progeny
pbmc@assays$progeny %>%dim()
pbmc@assays$progeny@data[,1:19]
# Assay data with 14 features for 2638 cells # First 10 features:
# Androgen, EGFR, Estrogen, Hypoxia, JAK-STAT, MAPK, NFkB, p53, PI3K, TGFb
3
## We can now directly apply Seurat functions in our Progeny scores.
## For instance, we scale the pathway activity scores.
pbmc <- Seurat::ScaleData(pbmc, assay = "progeny")
## We transform Progeny scores into a data frame to better handling the results
progeny_scores_df <- as.data.frame(
t(GetAssayData(pbmc, slot = "scale.data", assay = "progeny"))) %>%
rownames_to_column("Cell") %>%
gather(Pathway, Activity, -Cell)
dim(progeny_scores_df)
head(progeny_scores_df)
head( t(GetAssayData(pbmc, slot = "scale.data", assay = "progeny")))
head(CellsClusters)
# [1] 36932 3 ## We match Progeny scores with the cell clusters.
progeny_scores_df <- inner_join(progeny_scores_df,
CellsClusters)
head(progeny_scores_df)
## We summarize the Progeny scores by cellpopulation
summarized_progeny_scores <- progeny_scores_df %>%
group_by(Pathway, CellType) %>%
summarise(avg = mean(Activity), std = sd(Activity))
dim(summarized_progeny_scores)
head(summarized_progeny_scores)
# [1] 126 4 ## We prepare the data for the plot
summarized_progeny_scores_df <- summarized_progeny_scores %>%
dplyr::select(-std) %>%
spread(Pathway, avg) %>%
data.frame(row.names = 1, check.names = FALSE, stringsAsFactors = FALSE)
head(summarized_progeny_scores_df)
4#画图
paletteLength = 100
myColor = colorRampPalette(c("Darkblue", "white","red"))(paletteLength)
progenyBreaks = c(seq(min(summarized_progeny_scores_df), 0, length.out=ceiling(paletteLength/2) + 1),
seq(max(summarized_progeny_scores_df)/paletteLength,
max(summarized_progeny_scores_df), length.out=floor(paletteLength/2)))
progeny_hmap = pheatmap(t(summarized_progeny_scores_df),
fontsize=12,
fontsize_row = 10, color=myColor,
breaks = progenyBreaks, main = "PROGENy (500)",
angle_col = 45, treeheight_col = 0, border_color = NA)
#install.packages("viridis")
library(viridis)
DefaultAssay(pbmc) <- 'progeny'
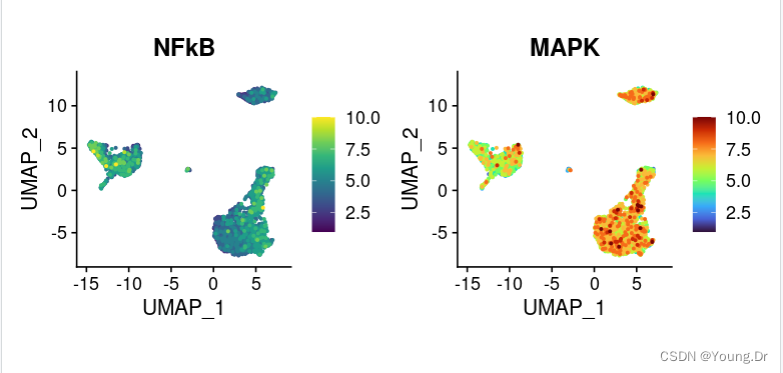
p1= FeaturePlot(pbmc,features = "NFkB", coord.fixed = T, order = T, cols = viridis(10))
p2=FeaturePlot(pbmc,features = "MAPK", coord.fixed = T, order = T, cols = viridis::turbo(10))
p1|p2