变异位点染色体分布图
今天分享的内容是通过RIdeogram包绘制染色体位点分布图,并介绍一种展示差异位点的方法。

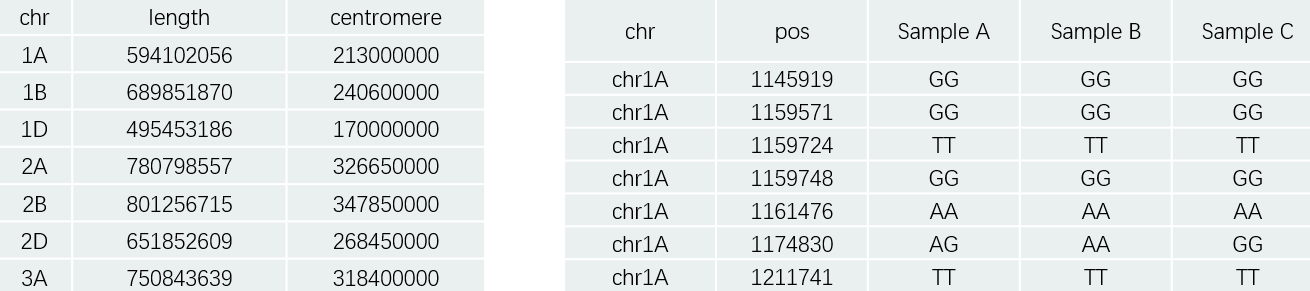
在遗传学研究中,通过测序等方式获得了基因组上某些位置的基因型信息。 如下表,第一列是变异位点的ID,第二列是染色体,第三列是物理位置,最后两列是两个样品的基因型。
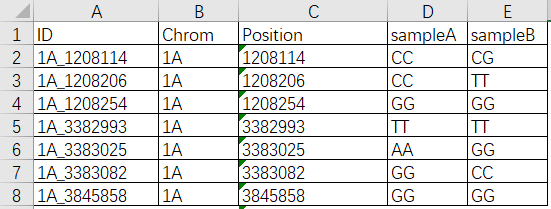
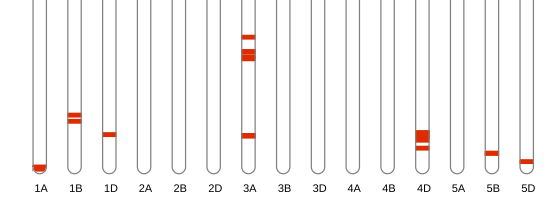
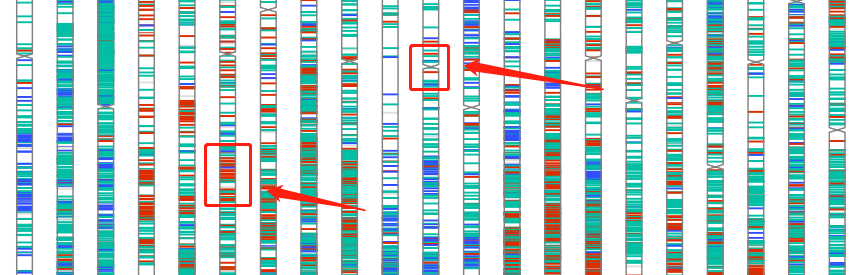
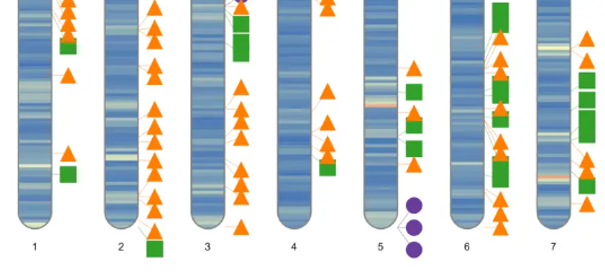
这个文件通常有好几万行,要想快速从中获得有效信息,可以借助变异位点染色体分布图功能实现。比如下图,红色部分表示样品A和样品B差异的位置,一眼就能看出哪些位置比较关键。
数据准备
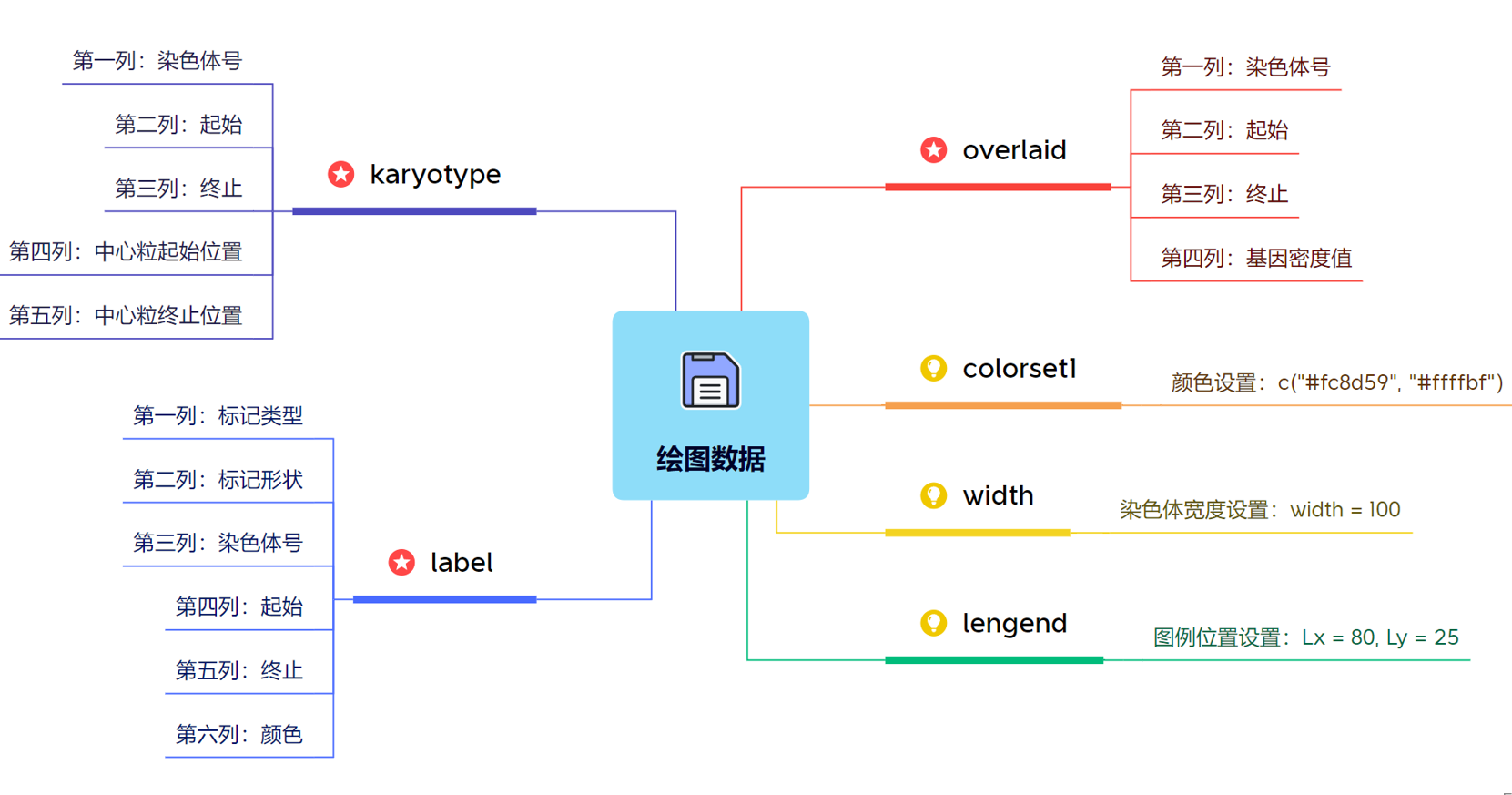
主要的输入文件有两个,第一个是染色体结构信息(长度、着丝粒位置等),第二个是待分析标注的位点信息(不同材料在某位置的基因型),如下所示:
操作方法
首先,安装并加载R包:
library(RIdeogram)
library(tidyverse)
library(xlsx)
设置参数
将整理好的文件保存为csv格式,输入文件名。centwidth是着丝粒显示宽度,singewidth是单个位点的显示宽度,这两个值可以自定义设置。(如果设置的值越大,那么线条就越粗)
my_filename <- "data.csv"
my_centwidth <- 5000000
my_color <- c("#e0e0e0", "#304ffe", "#dd2c00","#00bfa5")
my_singewidth <- 5000000
添加染色体信息
由于绘图时需要每个染色体的长度(起始和终止位置)和着丝粒的位置,因此提前准备一个txt文档,按如下格式整理数据,用于后续绘图。 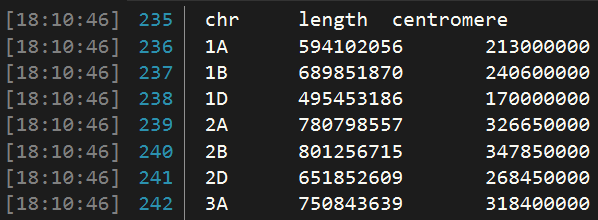
下面是读取染色体信息的代码:
df_chr_pos_cent <- read.table("./Ref_chromedata.txt",header = T)
df_chr_pos_cent <- cbind(df_chr_pos_cent$chr,"0",
df_chr_pos_cent$length,
df_chr_pos_cent$centromere,
as.numeric(df_chr_pos_cent$centromere) + my_centwidth)
df_chr_pos_cent <- as.data.frame(df_chr_pos_cent)
df_chr_pos_cent <- df_chr_pos_cent[1:21,] %>%
mutate_at(vars(V2,V3,V4,V5),as.integer)
colnames(df_chr_pos_cent) <- c("Chr","Start","End","CE_start","CE_end")
> head(df_chr_pos_cent)
Chr Start End CE_start CE_end
1 1A 0 594102056 213000000 218000000
2 1B 0 689851870 240600000 245600000
3 1D 0 495453186 170000000 175000000
4 2A 0 780798557 326650000 331650000
5 2B 0 801256715 347850000 352850000
6 2D 0 651852609 268450000 273450000
先从名为 "Ref_chromedata.txt" 的文件中读取数据,并将其存储在名为 df_chr_pos_cent 的数据框中,再选取某些列的内容,并将它们拼接成一个新的矩阵,自动计算每条染色体的起止位置和着丝粒的起止位置。
创建判定子代和亲本关系的函数
为了更方便的计算某个位点在亲本和子代中的存在情况,快速获得子代中突变信息的来源,创建一个亲缘关系判定函数。
# 函数:输入两个亲本和子代基因型,判断子代来源
determine_progeny <- function(p1,p2,prog,name_p1,name_p2){
if (p1 == p2){
if (p1 == prog & p2 == prog){
return("Both")
}else{
return("Unknown")
}
}else{
if (p1 == prog){
return(name_p1)
}else{
if (p2 == prog){
return(name_p2)
}else{
return("Unknown")
}
}
}
}
这个函数叫做 determine_progeny,它用于确定两个亲本(p1 和 p2)以及一个后代(prog)之间的关系。
参数说明:
-
p1 - 第一个亲本的标识。 -
p2 - 第二个亲本的标识。 -
prog - 后代的标识。 -
name_p1 - 第一个亲本的名字 -
name_p2 - 第二个亲本的名字
功能介绍:
首先,检查如果第一个亲本 p1 和第二个亲本 p2 相等,那么它们之间的关系有两种情况:
-
如果它们都等于后代 prog,那么返回 "Both",表示两个亲本都与后代相关。 -
另外一种情况,返回 "Unknown",表示无法确定它们之间的关系。
如果第一个亲本 p1 和第二个亲本 p2 不相等,那么它们之间的关系也有两种情况:
-
如果第一个亲本 p1 等于后代 prog,那么返回 name_p1,表示第一个亲本是后代的亲本。 -
如果第二个亲本 p2 等于后代 prog,那么返回 name_p2,表示第二个亲本是后代的亲本。
最后,如果以上条件都不满足,返回 "Unknown",表示无法确定它们之间的关系。 这个函数的目的是根据输入的亲本和后代信息,确定它们之间的关系,并返回一个相应的标注信息。
创建判定杂合子的函数
如果某些位点为杂合状态,需要对其进行识别标注,以下提供一种算法进行识别:
# 函数:判断杂合子
decide_hybrid <- function(genetype){
tem <- str_split(genetype,"")
if (length(tem[[1]]) != 2){
return("Error")
}else{
first <- tem[[1]][1]
sencend <- tem[[1]][2]
if (first == sencend){
return("equal")
}else{return("diff")}
}
}
这段代码定义了一个名为decide_hybrid的函数,其作用是接受一个字符串参数genetype,然后将该字符串拆分成两个字符,并检查这两个字符是否相等。
如果输入字符串的长度不等于2,函数将返回"Error",表示输入错误。
否则,如果两个字符相等,函数将返回"equal",表示相等。
如果两个字符不相等,函数将返回"diff",表示不相等。这个函数主要用于比较输入的基因型是否相等或不等。 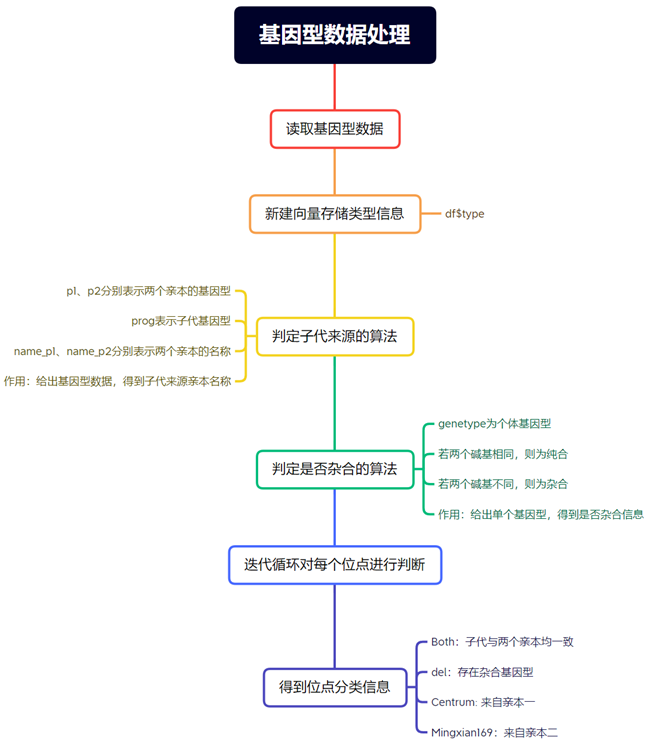
计算每个位点的变异类型
for (i in 1:nrow(df)){
if (df$chr[i] == "#N/A"){
df$type[i] <- "del"
next
}
if (df$pos[i] == "#N/A"){
df$type[i] <- "del"
next
}
if (decide_hybrid(df[i,3]) == "diff" |
decide_hybrid(df[i,4]) == "diff" |
decide_hybrid(df[i,5]) == "diff"){
df$type[i] <- "del"
next
}
df$type[i] <- determine_progeny(df[i,3],df[i,4],df[i,5],
colnames(df)[3],colnames(df)[4])
}
这段代码的主要作用是根据一系列条件来确定df 数据框中每一行的"type"列的值,并根据不同条件进行相应的处理。如果某些条件满足,它将类型设置为 "del",否则它会计算新的类型并将其存储在数据框中。
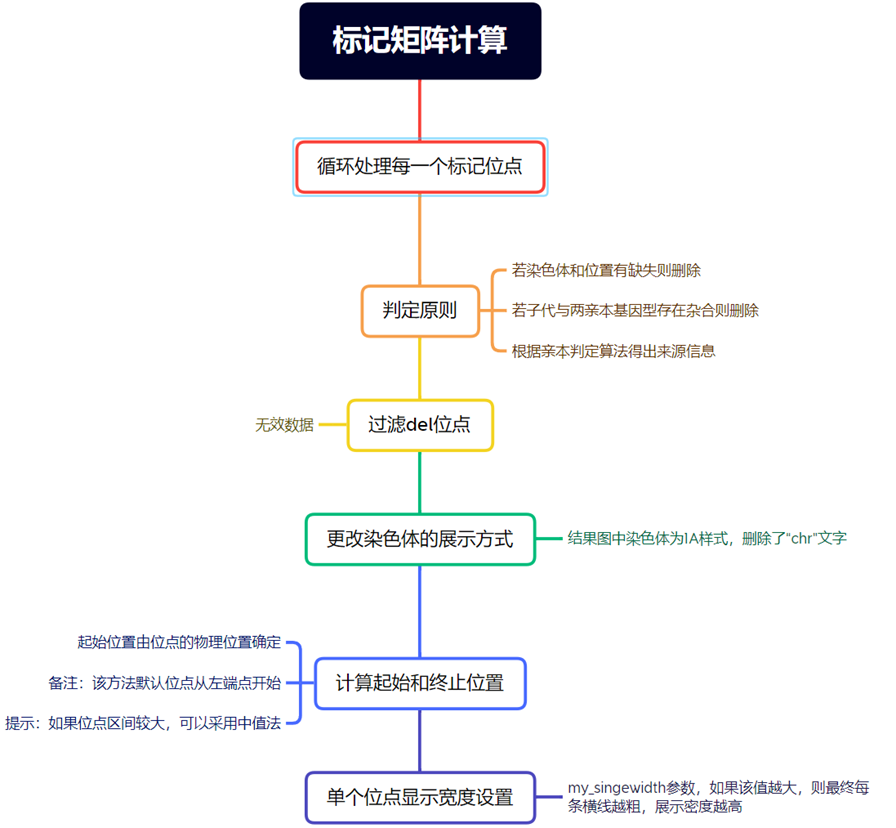
绘图数据初步整理
df_marker <- df %>% filter(type != "del")
df_marker$chr <- sub("chr","",df_marker$chr)
df_marker$Start <- df_marker$pos
df_marker$End <- as.numeric(df_marker$pos) + my_singewidth
df_marker$Value[which(df_marker$type == "Both")] <- 4
df_marker$Value[which(df_marker$type == colnames(df)[3])] <- 3
df_marker$Value[which(df_marker$type == colnames(df)[4])] <- 2
df_marker$Value[which(df_marker$type == "Unknown")] <- 1
首先从数据框df中筛选出type列不等于"del"的行,然后对筛选后的数据框df_marker进行一系列操作:
移除df_marker$chr列中的"chr"前缀,将结果存储回df_marker$chr中。 将df_marker$pos列的值赋给新的列df_marker$Start,以便将位置信息重命名。 根据my_singewidth的值计算新的列df_marker$End,该列的值等于df_marker$pos加上my_singewidth的数值。
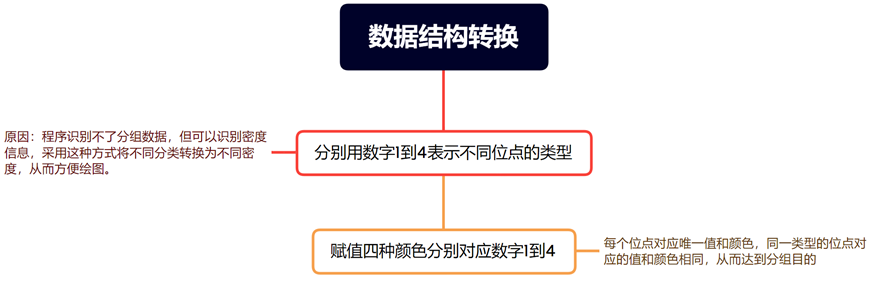
根据条件对df_marker$Value列进行更新,将根据type列的不同值分配不同的数值,以下为对应规则:
如果type等于"Both",则将df_marker$Value设置为4。
如果type等于数据框中的第三列的列名,则将df_marker$Value设置为3。
如果type等于数据框中的第四列的列名,则将df_marker$Value设置为2。
如果type等于"Unknown",则将df_marker$Value设置为1。
主要目的是根据条件对df数据框进行筛选和转换操作,创建一个数据框df_marker,其中包括重命名列、计算新列以及根据不同的type值赋予相应的数值。
转换数据格式
df_marker_plot <- cbind(df_marker[,c(1,7,8,9)])
df_marker_plot <- df_marker_plot %>%
mutate_at(vars(Start,End,Value),as.integer)
df_marker_plot <- df_marker_plot[which(df_marker_plot$chr !="Un"),]
colnames(df_marker_plot) <- c("Chr","Start","End","Value")
这段代码的功能是对名为df_marker的数据框进行处理,首先选择其中的特定列(第1、7、8、9列),然后将这些列的数据类型转换为整数类型,接着从中删除chr列值不等于"Un"的行,最后重命名这些列为"Chr"、"Start"、"End"和"Value",用于后续的数据可视化或分析。
绘制染色体全景图
# 绘制全景图
ideogram(karyotype = df_chr_pos_cent,
overlaid = df_marker_plot,
colorset1 = my_color)
convertSVG("chromosome.svg", device = "pdf")
最后,使用如上代码即可生成最终的结果图片。该R包非常巧妙的使用SVG语法构建图形,并且提供了svg转pdf、png等多种方式。
提示:分类型离散数据通过数值映射转换为连续型,便可以通过函数进行绘图计算,如果有多个维度的数据需要展示,可以添加其他类型的标注。
多组学应用:
-
差异表达基因的分布(RNA-seq) -
开放染色质的分布(ATAC-seq) -
CTCF结合位点(ChIP-seq) -
突变位点在染色体上的分布(WGS) -
DNA甲基化的分布(WGBS) -
外显子在染色体上的分布(WES)
遗传学应用:
-
变异位点全景图绘制 -
标记物理位置展示图绘制 -
遗传多样性来源展示 -
基因连锁定位结果展示
>>> 参考资料
https://github.com/cran/RIdeogram
http://doi.org/10.7717/peerj-cs.251
https://www.jianshu.com/p/07ae1fe18071
>>> Tips:本文所有示例数据均随机生成,不具有任何意义
本文由 mdnice 多平台发布



![[NLP] LLM---<训练中文LLama2(五)>对SFT后的LLama2进行DPO训练](https://img-blog.csdnimg.cn/51e0f89539834e8ca494b4d047d0a18c.png)
![[字符串和内存函数]错误信息报告函数strerror详解](https://img-blog.csdnimg.cn/98adee09ed56420f9f75783f0f52e2b8.jpeg)
![集成电路运算放大器[23-9-16]](https://img-blog.csdnimg.cn/943b7d0da5d541638f84ee604534edae.png)