目录
- 软件安装
- 新建文件夹
- 一、下载数据
- 二、质控过滤
- 1.数据质量检测
- 2.数据质量控制
- 3.对处理后的数据再次QC
- 三、序列比对
- 1.hisat2比对
- 2.flagstat检查一下结果
- 四、featureCounts定量
写在前面——本文是转录组上游分析的实战练习。主要包含四个步骤:
- 数据下载(包括sra数据、参考基因组、参考基因组注释文件)
- 质控过滤(使用trim-galore进行质控,使用fastqc、multiqc进行质量检测)
- 序列比对(hisat2)
- featureCounts定量
软件安装
详细步骤见
RNA-seq——一、Linux软件安装
安装配置conda
wget -c https://mirrors.bfsu.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
source ~/.bashrc
conda config --add channels r
conda config --add channels bioconda
conda config --add channels conda-forge
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.bfsu.edu.cn/anaconda/cloud/bioconda/
conda config --set show_channel_urls yes
使用conda安装软件
conda create -n rna python=3.8
conda activate rna
conda install fastqc
conda install multiqc
conda install trim-galore
conda install hisat2
conda install samtools
conda install subread
新建文件夹
00ref:存放参考基因组及注释文件
01raw_data:存放原始数据
02clean_data:存放清洗之后的数据
03align_data:存放比对后的文件
04matrix:存放reads计数文件
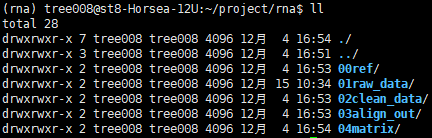
文件结构清晰,让人赏心悦目~
注:练习时注意当前所在位置,要在正确的文件夹中进行正确的操作。
一、下载数据
conda install sra-tools
conda update sra-tools
# -p Show progress
prefetch -p SRR11618610
prefetch -p SRR11618616
prefetch -p SRR11618621
检查数据是否完整
md5sum *.sra > md5.txt
cat md5.txt
md5sum -c md5.txt

sra数据处理:
fastq-dump
优点:可以直接将sra文件转为fastq.gz文件
缺点:不能自定义线程
fasterq-dump
优点:可自定义线程,面对大量数据时,效率更高
缺点:sra转为fastq,再压缩成fastq.gz
其他工具:kingfisher
Kingfisher是一个高通量测序数据下载工具,能根据用户的需求将下载数据直接输出为SRA、Fastq、Fasta或Gzip等格式,非常方便,不需要自己再对SRA数据通过fasterq-dump进行拆分转换。
fastq-dump --gzip --split-3 SRR*.sra
此处数据较少,偷个懒~
# 查看数据
zcat SRR11618610_1.fastq.gz | head -n 8
最终结果如图:

二、质控过滤
1.数据质量检测
# 分别对单个文件进行检测,输出多个html格式的检测结果
fastqc SRR*gz
# 整合检测结果
multiqc ./
检测结果(MultiQC Report)主要包含以下内容:
- Sequence Quality Histograms
- Per Sequence Quality Scores
- Per Base Sequence Content
- Per Sequence GC Content
- Per Base N Content
- Sequence Length Distribution
- Sequence Duplication Levels
- Overrepresented sequences
- Adapter Content
- Status Checks
经过检测,这三个数据集存在一些问题,具体如下:
Per Base Sequence Content

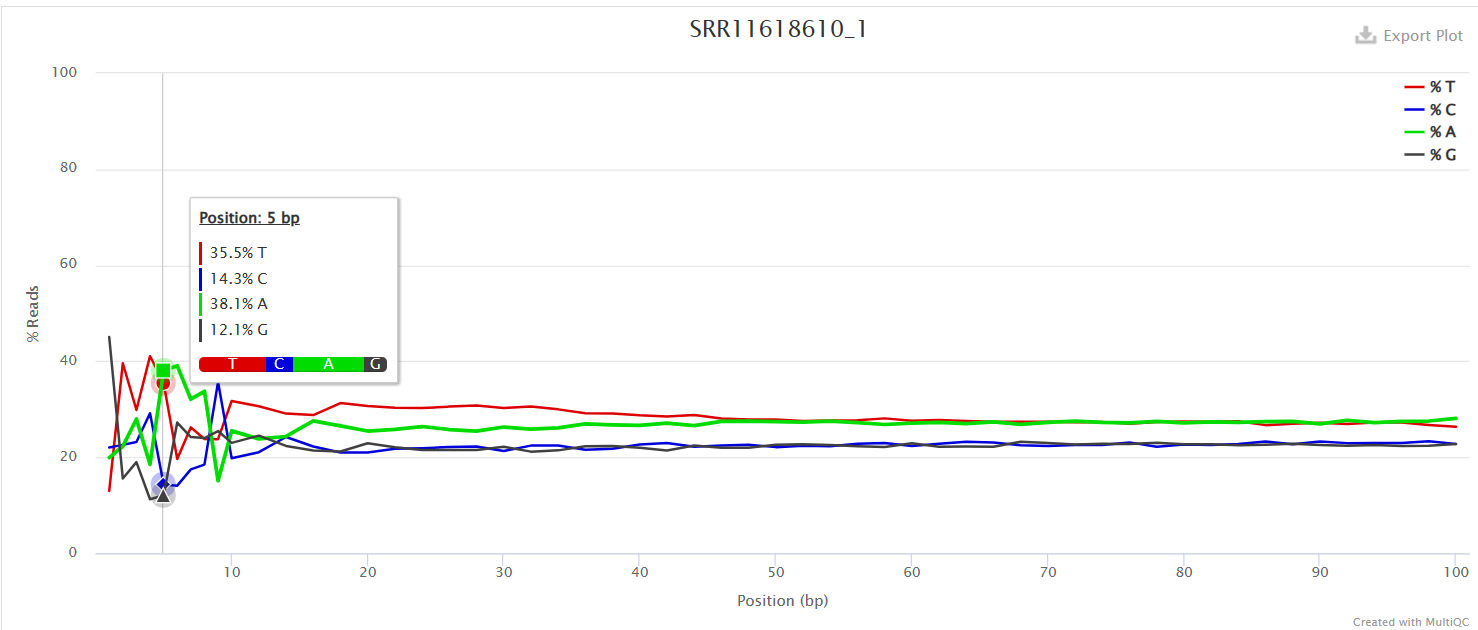
对所有reads的每一个位置,统计ATCG四种碱基的分布,正常情况下每个位置每种碱基出现的概率是相近的,四条线应该平行且相近。
当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。
本结果前10个位置,每种碱基频率有明显的差别,说明有污染。
当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
Per Sequence GC Content

统计reads的平均GC含量的分布。理论分布应该是正态分布,均值不一定在50%,而是由平均GC含量推断的。
曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。
形状接近正态但偏离理论分布的情况提示可能有系统偏差。偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
Sequence Duplication Levels

统计不同拷贝数的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在。
图中显示大于10个重复的reads占总序列的20%以上。
当非unique的reads占总数的比例大于20%时,报"WARN";当非unique的reads占总数的比例大于50%时,报"FAIL“。
2.数据质量控制
FASTQ文件格式及测序文件phred质量格式判断

判断一下测序文件phred的格式,为了之后选择trim_galore的参数。
目前主流的格式为Phred+33
vim fa_type.sh
# 脚本中加入如下内容
# 编辑完成之后 wq 保存
less $1 | head -n 1000 | awk '{if(NR%4==0) printf("%s",$0);}' | od -A n -t u1 -v | awk 'BEGIN{min=100;max=0;} \
{for(i=1;i<=NF;i++) {if($i>max) max=$i; if($i<min) min=$i;}} \
END{if(max<=126 && min<59) print "Phred33"; \
else if(max>73 && min>=64) print "Phred64"; \
else if(min>=59 && min<64 && max>73) print "Solexa64"; \
else print "Unknown score encoding";}'
# 运行脚本
bash fa_type.sh SRR11618610_1.fastq.gz
# 输出结果
Phred33
使用trim_galore批量去除adapter、过滤掉低质量的reads
参考:5 trim_galore去接头(并行处理)
# 文件分类
ls | grep _1.fastq.gz > gz1
ls | grep _2.fastq.gz > gz2
paste gz1 gz2 > config
vim trim.sh
# trim.sh中的代码
dir=/home/st8/ssd2/tree008/project/rna/02clean_data/
cat config |while read id
do
arr=${id}
fq1=${arr[0]}
fq2=${arr[1]}
nohup trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o $dir $fq1 $fq2 &
done
# 运行脚本
bash trim.sh

参数说明:
-q/–quality
移除接头,修剪3’端低质量的碱基。默认值为20。
–phred33
适用于IlLumina 1.9+:指导cutadapt使用ASCII+33质量分数作为pared分数,默认使用。
–stringency
接头序列最小配对碱基数:简单来说就是最多能允许末端残留多少个接头序列的碱基,默认值为极端值1。
–length
设置长度阈值,若read通过质控清洗或去接头后长度小于此阈值,则会被剔除。
对于双端结果,一对reads中若一个read因为该原因被抛弃,则对应的另一个read也抛弃。不会被输出到双端结果文件。
默认值:20bp。
–paired
对于双端结果,一对reads中若一个read因为质量或其他原因被抛弃,则对应的另一个read也抛弃,但若使用–retain_unpaired选项可以保留。
处理后的文件
# -d:分隔符,按照指定分隔符分割列。与-f一起使用
# -f:依据-d的分隔字符将一段信息分割成为数段,用-f取出第几段的意思
ls -lh *fq.gz | cut -d" " -f 5-

3.对处理后的数据再次QC
fastqc SRR*gz
multiqc ./
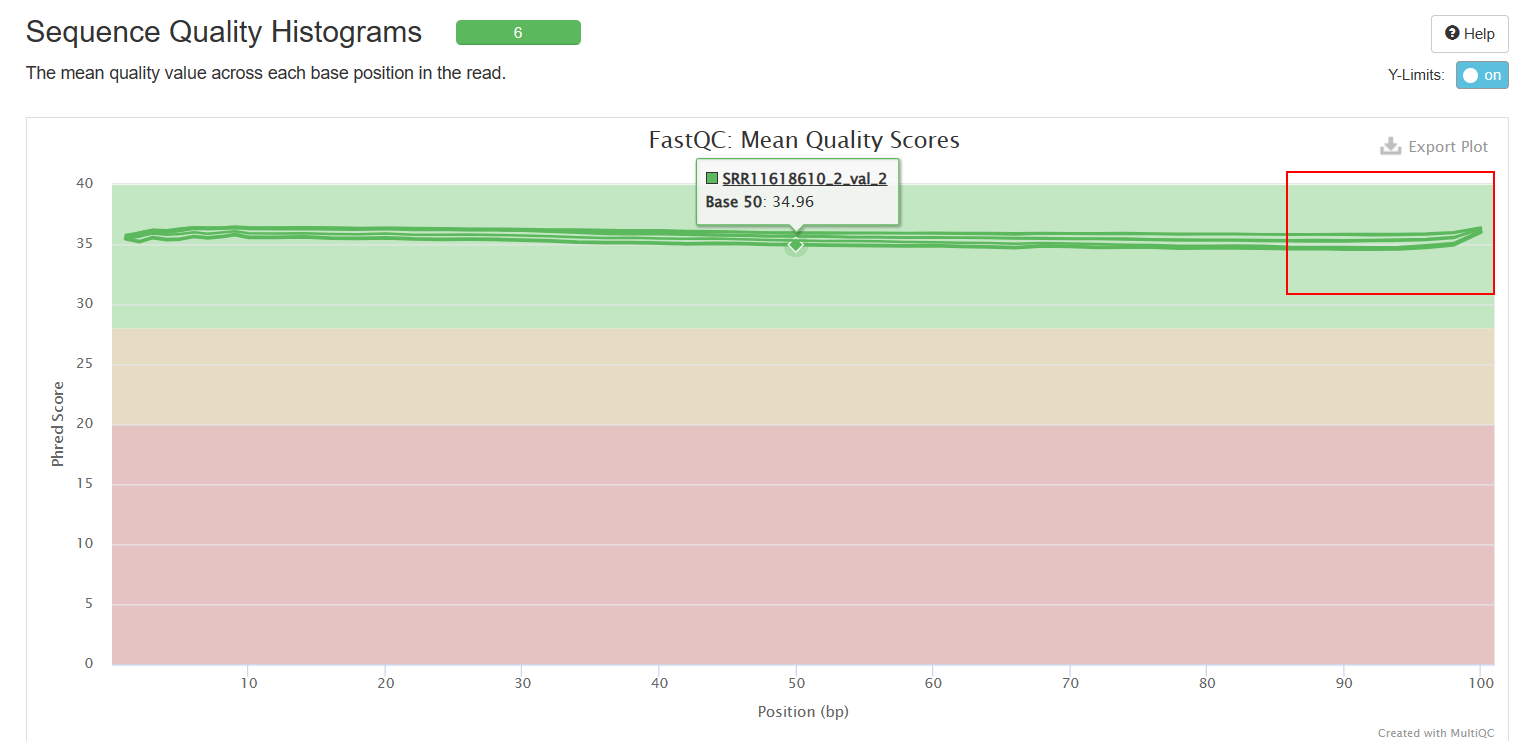
Sequence Quality Histograms
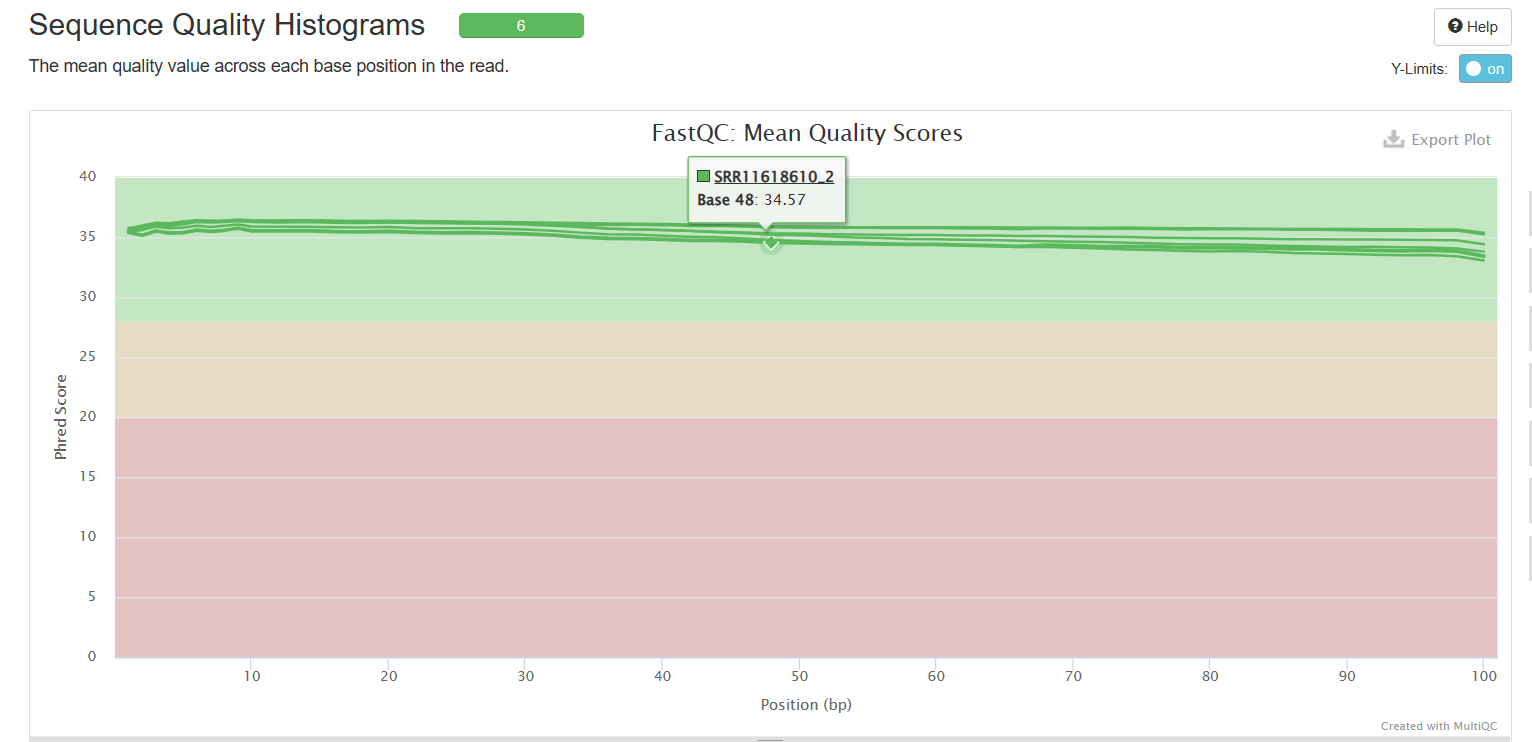
Adapter Content
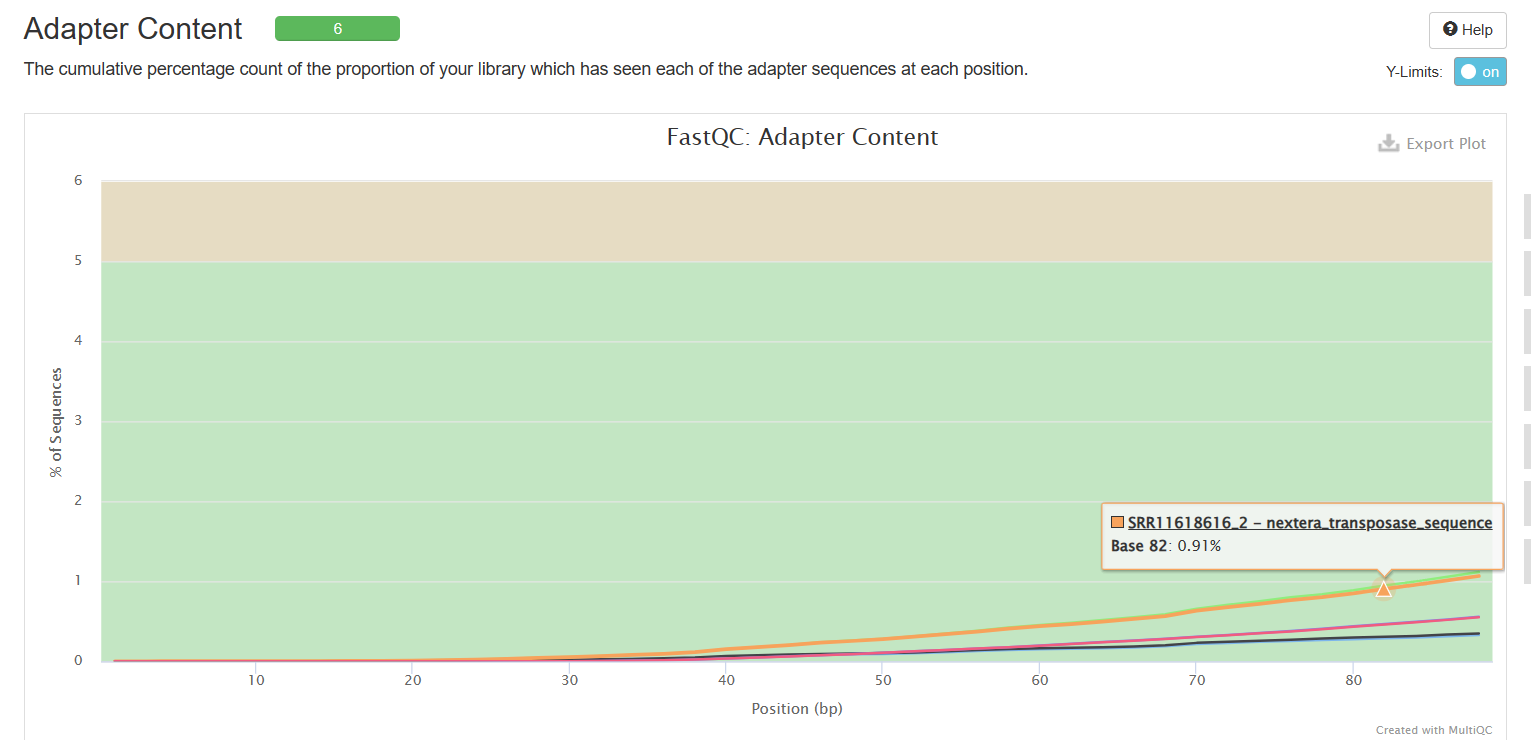
可以看到经过trim_galore处理之后,序列质量得到了提升(初始数据质量很好,所以提升不太明显),adapter也被去除。
三、序列比对
使用hisat2进行序列比对,需要先下载index。下载地址:https://daehwankimlab.github.io/hisat2/download/

genome: HISAT2 index for reference
genome_snp: HISAT2 Graph index for reference plus SNPs
genome_tran: HISAT2 Graph index for reference plus transcripts
genome_snp_tran: HISAT2 Graph index for reference plus SNPs and transcripts
# 这里我直接从别人那里cp了一份
cp /tmp/grch38_genome.tar.gz project/rna/00ref/
tar -zxvf grch38_genome.tar.gz

1.hisat2比对
ls *fq.gz | cut -d "_" -f 1 |
while read id; do nohup sh -c
"hisat2 -p 10 -x ../00ref/grch38/genome
-1 ${id}_1_val_1.fq.gz
-2 ${id}_2_val_2.fq.gz 2>${id}.log |
samtools sort -@ 10 -o ../03align_out/${id}.bam" & done
参数说明:
sh [参数] 脚本
-c 命令从字符串读取
-i 实现脚本交互
-n 进行语法检查
-x 实现逐条语句的跟踪
hisat2 [参数]
-p 设置线程
-x 参考基因组索引文件的前缀
-1 -2 分别对应双端测序的两个文件
samtools [参数]
sort 默认按照染色体位置进行排序
-@ 设置线程,加快运行速度
-o 设置最终排序后的输出文件名
其中,2>${id}.log是以覆盖的方式,把前面指令的错误信息输出到log文件中。好处就是把命令的结果保存起来,当我们需要的时候可以随时查询。具体见:
Linux 命令行shell输出重定向使用说明
Linux命令 结果输出重定向
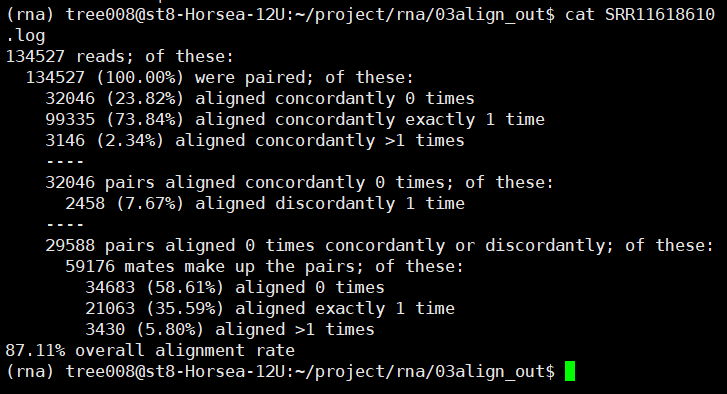
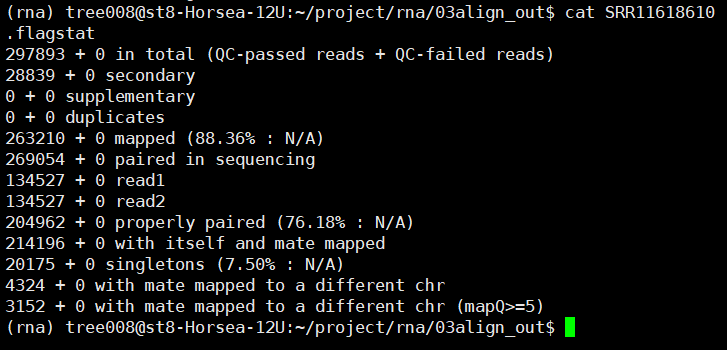
2.flagstat检查一下结果
ls *.bam | while read id;
do nohup samtools flagstat
-@ 10 ${id%.*}.bam > ${id%.*}.flagstat & done
##和%%表示最长匹配,#和%表示最短匹配。#是对左边部分处理,%是对右边部分处理。例子见:
https://baijiahao.baidu.com/s?id=1701830551588131996
四、featureCounts定量
定量需要gtf文件(参考基因组注释文件),下载地址:
https://www.gencodegenes.org/human/

gunzip gencode.v42.annotation.gtf.gz
gtf=/home/st8/ssd2/tree008/project/rna/00ref/gencode.v42.annotation.gtf
# 操作时位于03align_out文件夹
nohup featureCounts -T 10 -p -t exon
-g gene_id -a $gtf -o ../04matrix/all.id.txt
*.bam 1>../04matrix/counts.id.log 2>&1 &
参数说明
-T 线程数量,默认为1
-p 只能用在paired-end(双端测序)的情况中 If specified, fragments (or templates) will be counted instead of reads. This option is only applicable for paired-end reads; single-end reads are always counted as reads.
-t 设置feature-type,-t指定的必须是gtf中有的feature,同时read只有落到这些feature上才会被统计到,默认是“exon”
-g 在GTF注释中指定属性类型。默认为“gene_id”
-a 注释文件名称。默认为GTF/GFF格式
-o 输出文件的名称,包括read counts
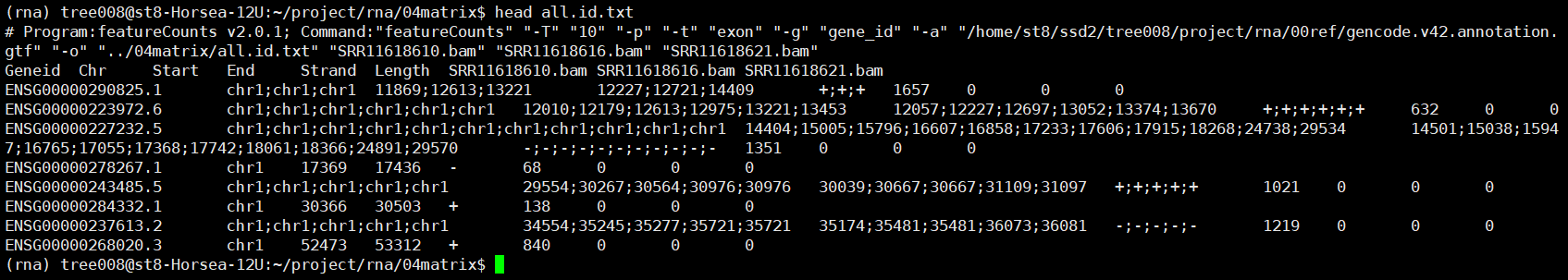
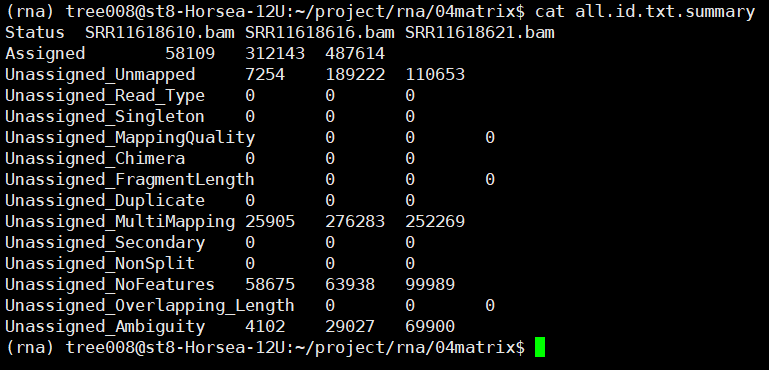
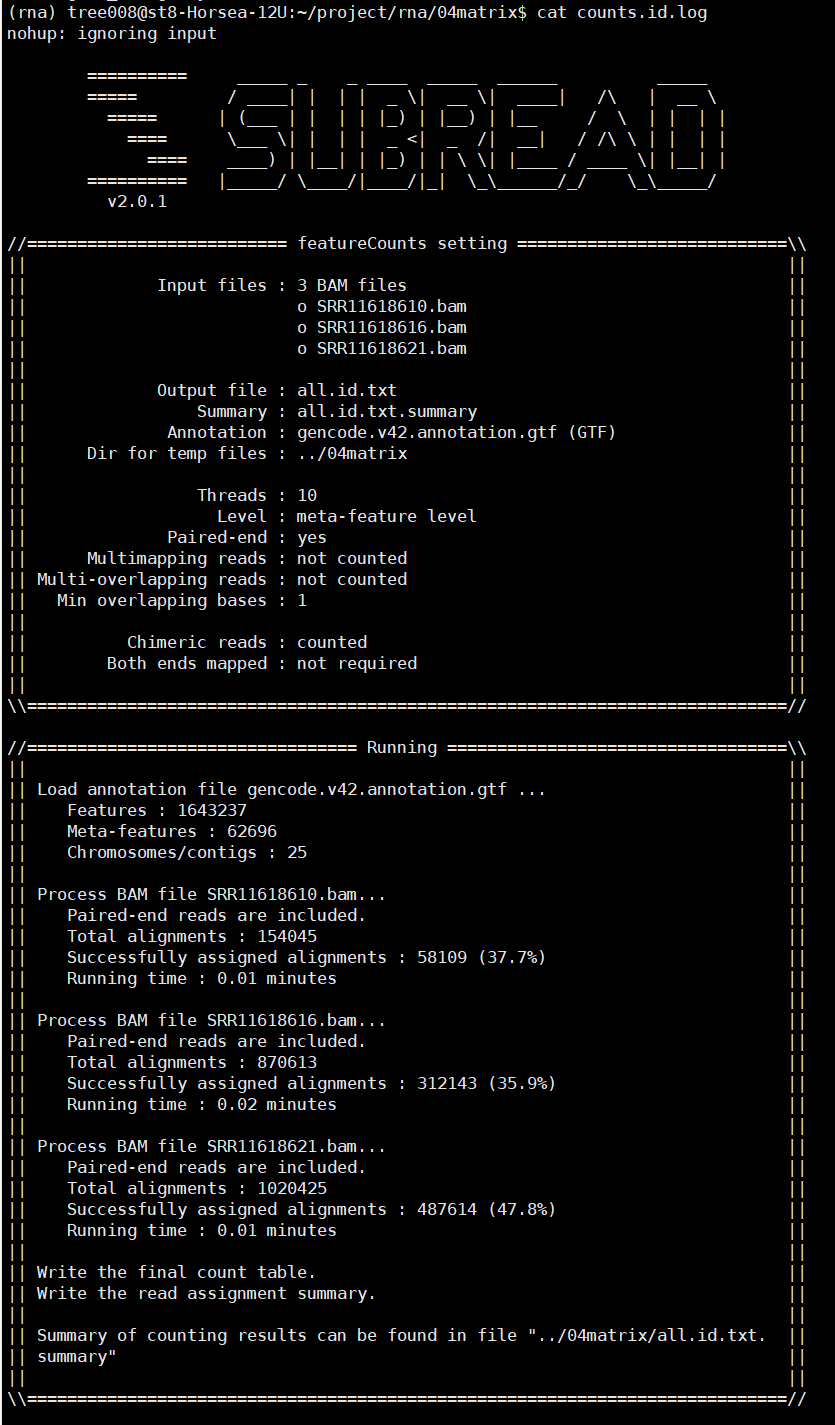
之后可以使用Rstudio对all.id.txt文件进行操作,上游分析到此为止。



![[附源码]Python计算机毕业设计Django网约车智能接单规划小程序](https://img-blog.csdnimg.cn/be2732ba41514c5ea1da6586f775e0b2.png)
![[附源码]Python计算机毕业设计华夏商场红酒管理系统Django(程序+LW)](https://img-blog.csdnimg.cn/706cd586d3824354b3644c319624066a.png)