关键词:相互作用 MS Forcite 分子动力学 径向分布 笔名:杨过
Forcite模块是分子动力学计算的主要模块,研究范围广,可以对多种周期性体系进行计算分析,在矿物分选领域中主要是计算分析药剂与矿物相互作用,在不同计算参数条件下可以实现药剂与矿物相互作用模型的预测与分析,从而得到表面相互作用机理。
因此,本文主要讲述运用Forcite模块对药剂与矿物相互作用计算过程分析。选取氯化胆碱-丙二酸(1:2)作为药剂,矿物选取氧化锌,对其进行模型搭建与计算。
首先将计算表面能得到的氧化锌(001)面完全解理面进行扩胞,建立6×6×4超胞模型,并运用Castep模块进行优化计算,然后通过Build layers将优化好的氯化胆碱-丙二酸(1:2)添加到已经扩胞优化好的氧化锌(001)超胞表面,并添加一定的真空层厚度避免周期性边界条件下力场的重复干扰。对搭建得到的模型进行几何结构优化,通过不断优化确定了最优的力场参数为CompassⅡ,选择Forcefield assigned电荷分布方法,Smart优化计算方法。进行分子动力学计算时选择NVT系综,温度控制选择NHL,求解牛顿运动方程应用Velocity Verlet 算法,静电力描述选择Ewald 方法,范德华作用力求解选择Atom-based 方法,截断半径为9.5 Å。总模拟时间为 1500 ps,每一步骤时间为 1 fs,总的模拟步骤为 1500000,最终得到稳定的相互作用体系并对其相互作用机理进行计算分析。
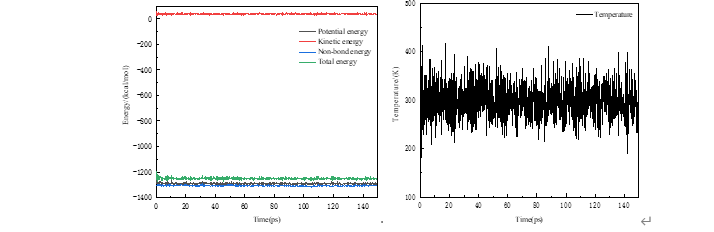
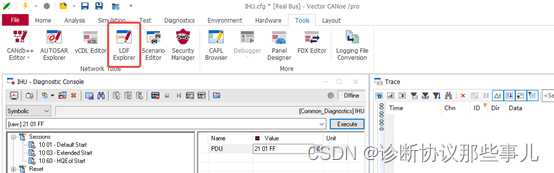
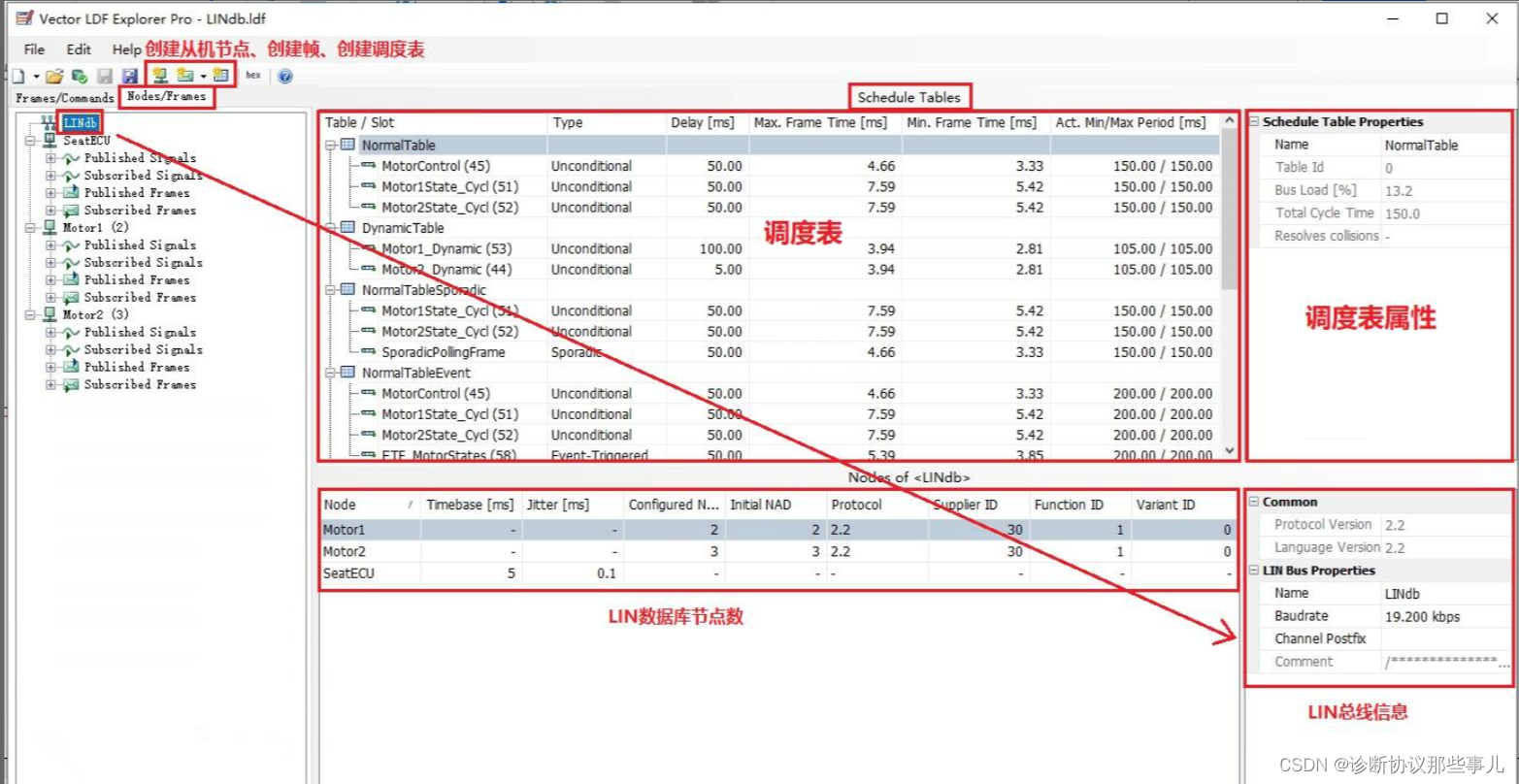
ChCl-2MA在ZnO(001)面动力学模拟(左:能量变化 右:温度变化)
由上图可以看出, ChCl-2MA与氧化锌在进行NVT系综模拟之后,分子间各种能量不断变化,最终氧化锌与ChCl-2MA相互作用体系相互作用能量在一定范围内趋于稳定。同时,ChCl-2MA与氧化锌分子动力学模拟过程中温度在300 K范围内变化, 表明相互作用体系温度变化已经趋于稳定。
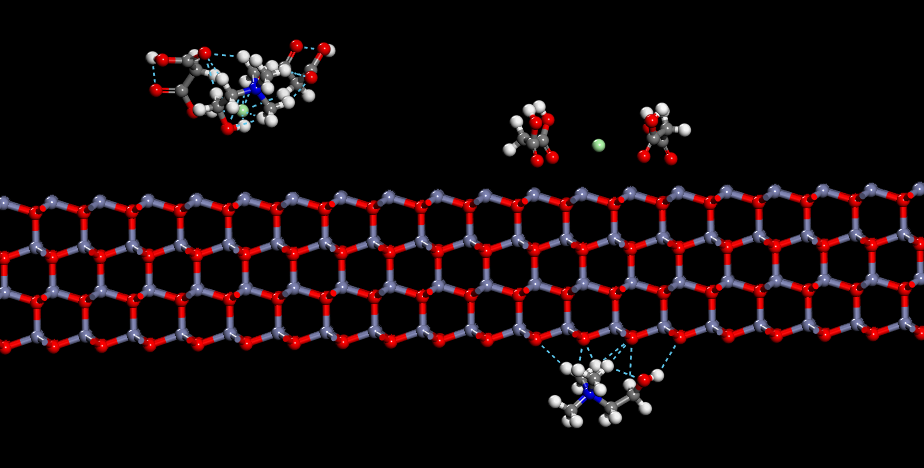
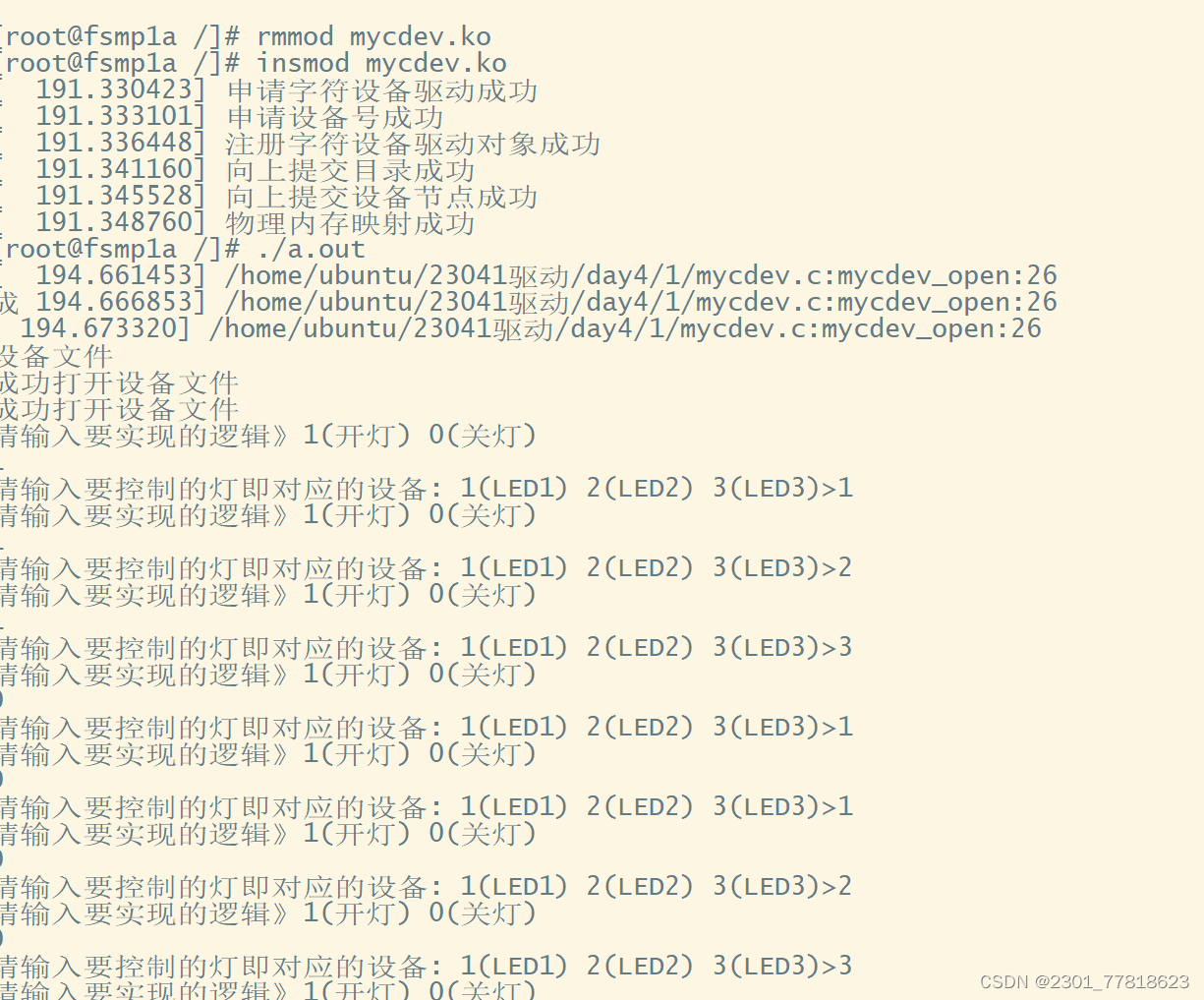
ChCl-2MA与ZnO(001)面吸附模型(左:相互作用前 右:相互作用后)
由上图可以看出ChCl-2MA在ZnO(001)上下表面发生相互作用。通过多重氢键相结合的ChCl-2MA低共熔溶剂在与ZnO(001)面相互作用过程中发生分解。其中氯化胆碱中的Cl和氢键供体作用在Zn突出表面,氯化胆碱中的胆碱阳离子作用在ZnO(001)面中O突出表面,胆碱阳离子中的部分C-H键与氧化锌表面上的O形成多重氢键吸附在氧化锌表面。
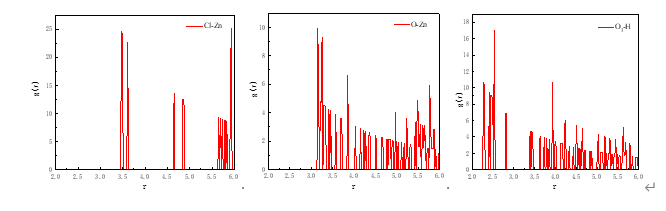
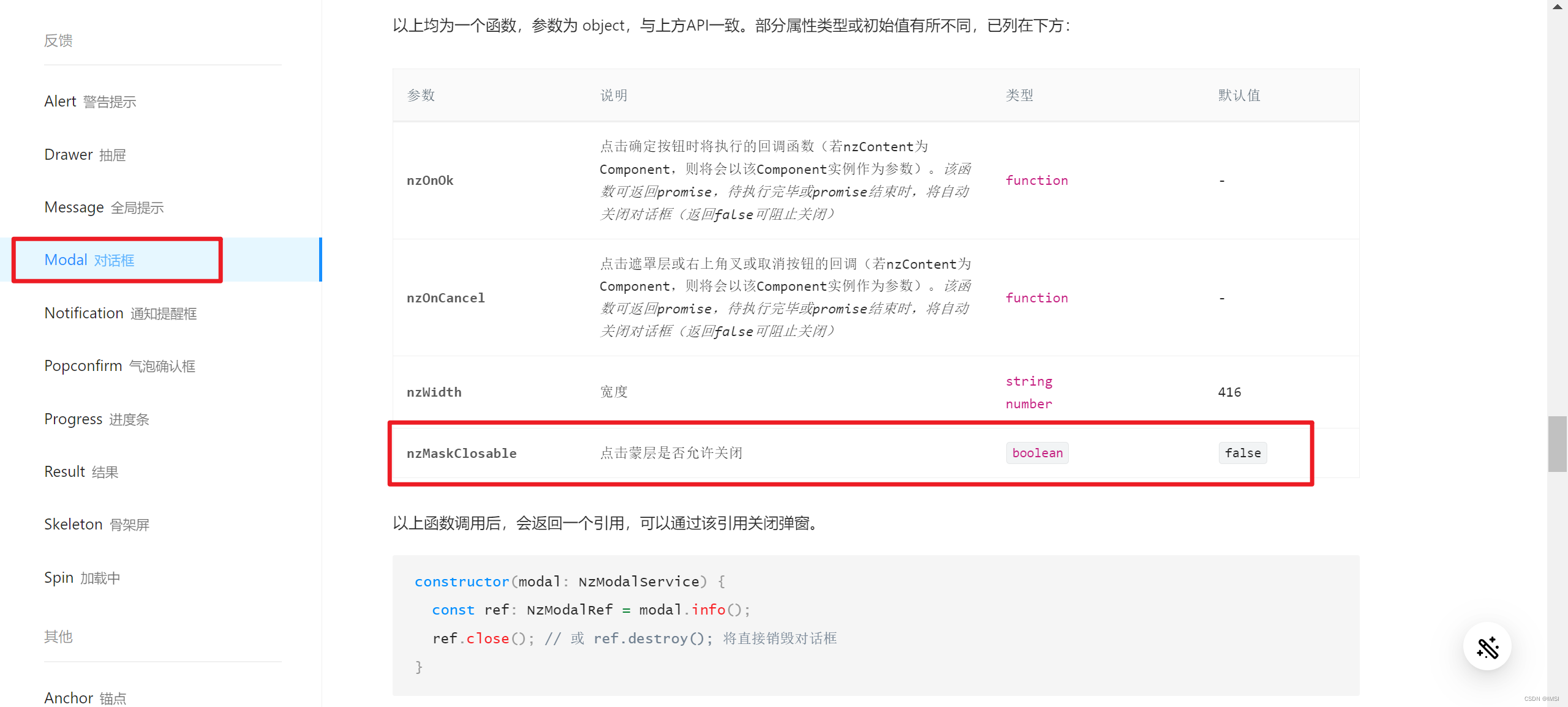
ZnO(001)面与ChCl-2MA径向分布图
由上图可以得出ChCl-2MA低共熔溶剂与氧化锌相互作用共分为三部分, ChCl-2MA中的Cl和C=O中的O与氧化锌中的Zn之间的距离在3~3.5内并且介于两种原子的共价键半径之和之间,表明Cl、O与Zn以化学作用的形式相互结合。而氧化锌中的氧原子与两种羧酸类低共熔溶剂中胆碱阳离子上的C-H、O-H以氢键方式相结合。通过径向分布图中各种相互作用的峰值可以看出Cl与Zn之间的相互作用占据主导地位,O…H次之,O-Zn最弱。由此说明氧化锌与ChCl-2MA相互作用形式既有化学作用又有物理作用,其中化学作用强度大于物理作用。
最后,有相关需求欢迎通过公众号"320科技工作室"联系