之前我们写过GO、KEGG的富集分析,参见:补充更新:GO、KEGG(批量分组)分析及可视化。演示了差异基因KEGG或者GO的分析流程。其实差异基因的富集分析输入的文件只需要一组基因就可以了。所以我们发挥了专治懒病的优良传统,将KEGG、GO(BP、CC、MF)的分析封装为一个函数,您只需要提供gene,选择物种即可,只有human和mouse。而且一次性完成KEGG和GO分析结果,免去了分析两次的麻烦。这样应该也不会出错了吧。
函数内容如下:其中相关参数可按照自己的需求修改!
Enrichment_KEGGgo_analusis <- function(genes,species=c('human','mouse')){library(org.Hs.eg.db)library(clusterProfiler)if(species == 'human'){genes_df <- bitr(genes,fromType="SYMBOL",toType="ENTREZID",OrgDb="org.Hs.eg.db",drop = TRUE)organism = "hsa"OrgDb = org.Hs.eg.db}if(species == 'mouse'){genes_df <- bitr(genes,fromType="SYMBOL",toType="ENTREZID",OrgDb="org.Mm.eg.db",drop = TRUE)organism = "mmu"OrgDb = org.Mm.eg.db}colnames(genes_df) <- c("gene","EntrzID")# KEGGkegg.re <- enrichKEGG(gene = genes_df$EntrzID,organism = organism,keyType = "kegg",pAdjustMethod = "fdr",pvalueCutoff = 0.05,qvalueCutoff = 0.05,minGSSize = 10,maxGSSize = 500)if (is.null(kegg.re)) {} else {kegg.re <- setReadable(kegg.re, OrgDb = OrgDb, keyType="ENTREZID")}print("kegg Done")# GOgo.re1 <- enrichGO(gene = genes_df$EntrzID,keyType = "ENTREZID",OrgDb= OrgDb,ont="BP",pAdjustMethod = "fdr",pvalueCutoff = 0.05,qvalueCutoff = 0.05,minGSSize = 10,maxGSSize = 500,readable = TRUE);print("GOBP Done")go.re2 <- enrichGO(gene = genes_df$EntrzID,keyType = "ENTREZID",OrgDb= OrgDb,ont="CC",pAdjustMethod = "fdr",pvalueCutoff = 0.05,qvalueCutoff = 0.05,minGSSize = 10,maxGSSize = 500,readable = TRUE);print("GOCC Done")go.re3 <- enrichGO(gene = genes_df$EntrzID,keyType = "ENTREZID",OrgDb= OrgDb,ont="MF",pAdjustMethod = "fdr",pvalueCutoff = 0.05,qvalueCutoff = 0.05,minGSSize = 10,maxGSSize = 500,readable = TRUE);print("GOMF Done")enrich_list <- list(kegg.re, go.re1, go.re2, go.re3)names(enrich_list) <- c("KEGG","GO_BP","GO_CC","GO_MF")return(enrich_list)}
我们演示一下。这里我们直接用向量提供了基因。如果您的文件是差异基因,很好弄,只需要$符号传入gene symbol那一列即可。
genes <- c(c('MAST4','IL4R','SYT1','PRDM1','AUTS2','KNL1','CD79A', "PLXDC2","NKG7","NELL2","BACH2","DIAPH3","SYN3", "NTNG1", "ADAM23","SOX5","TMPO","ARHGAP6","FCRL1","CD19"))results <- Enrichment_KEGGgo_analusis(genes = genes,species = 'human')#运行日志载入需要的程辑包:AnnotationDbiclusterProfiler v4.6.2 For help: https://yulab-smu.top/biomedical-knowledge-mining-book/If you use clusterProfiler in published research, please cite:T Wu, E Hu, S Xu, M Chen, P Guo, Z Dai, T Feng, L Zhou, W Tang, L Zhan, X Fu, S Liu, X Bo, and G Yu. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021, 2(3):100141载入程辑包:‘clusterProfiler’The following object is masked from ‘package:AnnotationDbi’:selectThe following object is masked from ‘package:IRanges’:sliceThe following object is masked from ‘package:S4Vectors’:renameThe following objects are masked from ‘package:plyr’:arrange, mutate, rename, summariseThe following object is masked from ‘package:stats’:filter'select()' returned 1:1 mapping between keys and columnsReading KEGG annotation online: "https://rest.kegg.jp/link/hsa/pathway"...Reading KEGG annotation online: "https://rest.kegg.jp/list/pathway/hsa"...[1] "kegg Done"[1] "GOBP Done"[1] "GOCC Done"[1] "GOMF Done"Warning messages:1: 程辑包‘AnnotationDbi’是用R版本4.2.2 来建造的2: In utils::download.file(url, quiet = TRUE, method = method, ...) :the 'wininet' method is deprecated for http:// and https:// URLs3: In utils::download.file(url, quiet = TRUE, method = method, ...) :the 'wininet' method is deprecated for http:// and https:// URLs
结果分别储存在list中,这样很方便了吧!
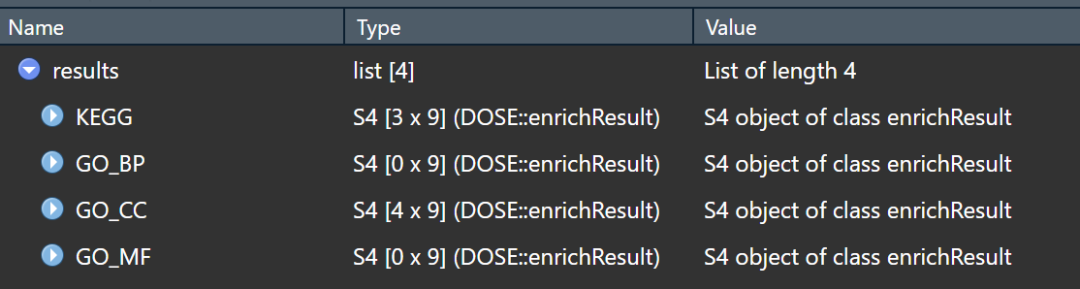
有需要的可以试一下,总之是为了省时省力,那些在线的分析工具的底层原理也就是这样。觉得分享有用的点个赞、分享下再走呗!