为什么介绍lotus2
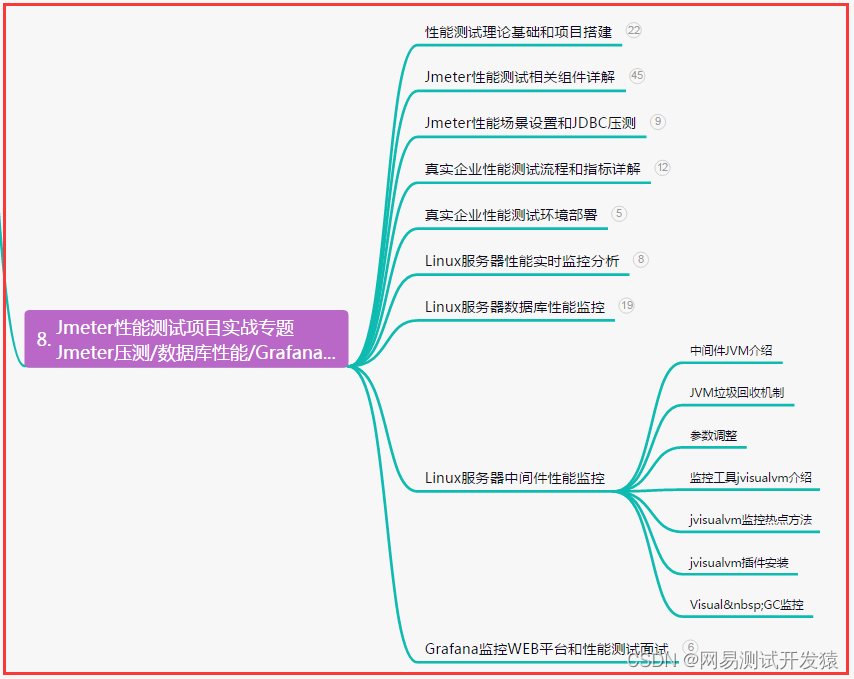
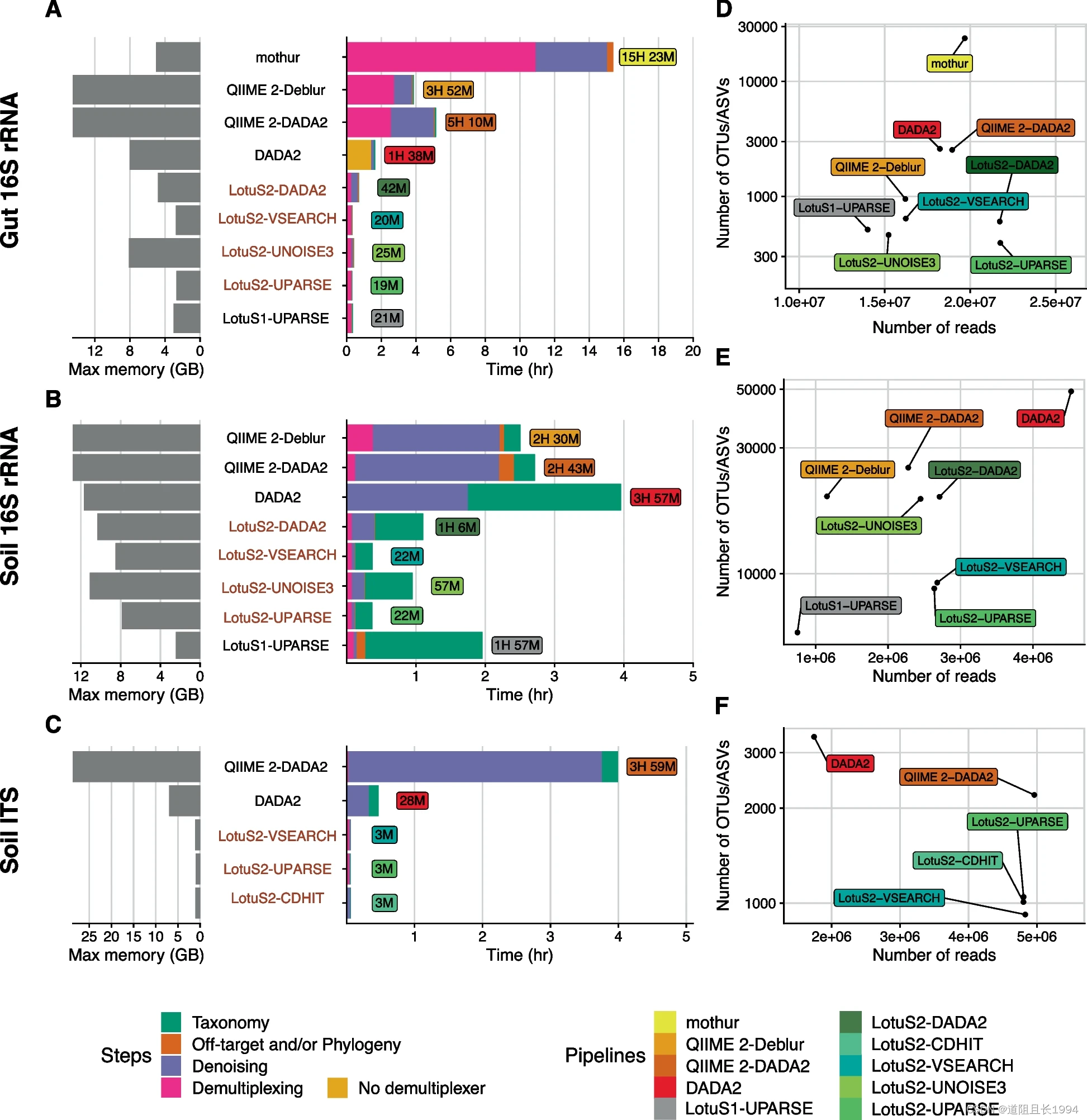
- 因为快,作者比较了lotus2流程和qiime2、dada2、vsearch等,lotus2的速度最快、占用内存最小。
- 因为方便,只需要一行代码,即可完成全部分析。
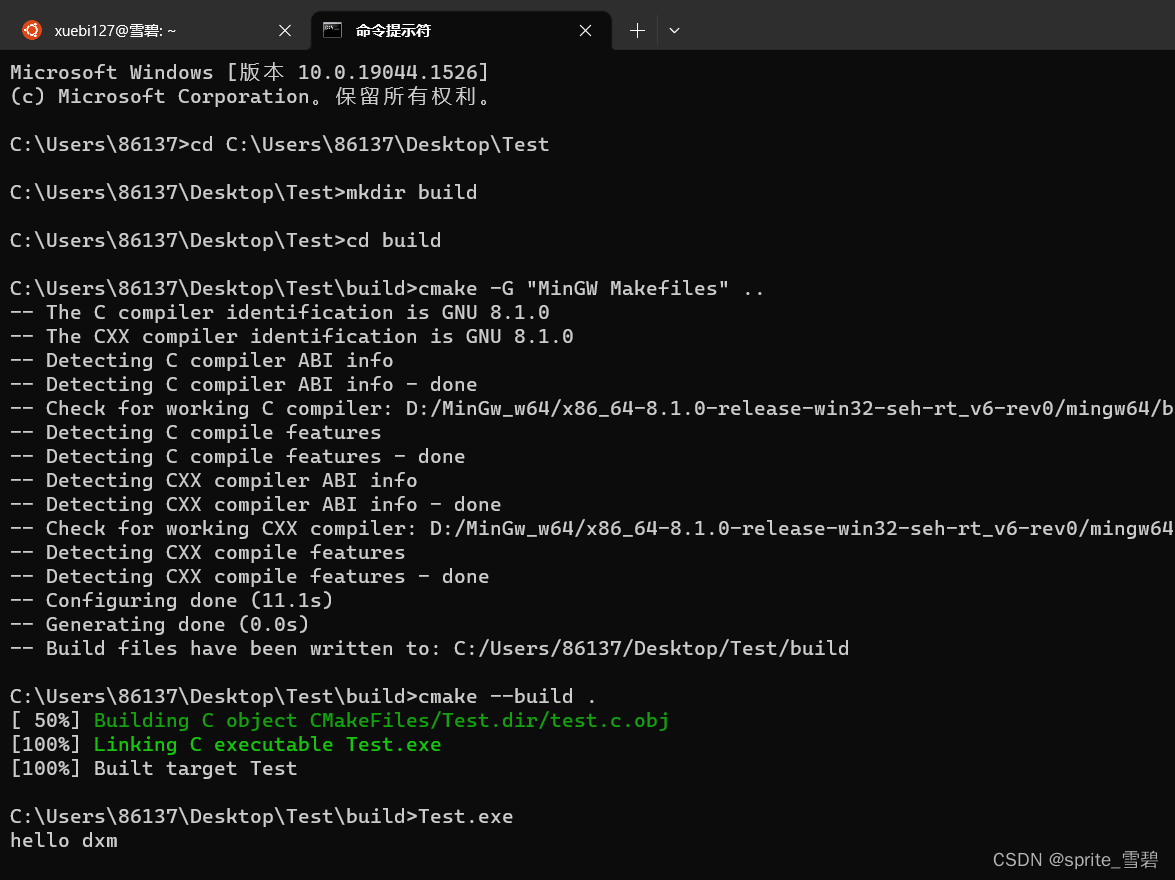
lotus2 -i Example/ -m Example/miSeqMap.sm.txt -o myTestRun
- 而且分类注释更加准确。
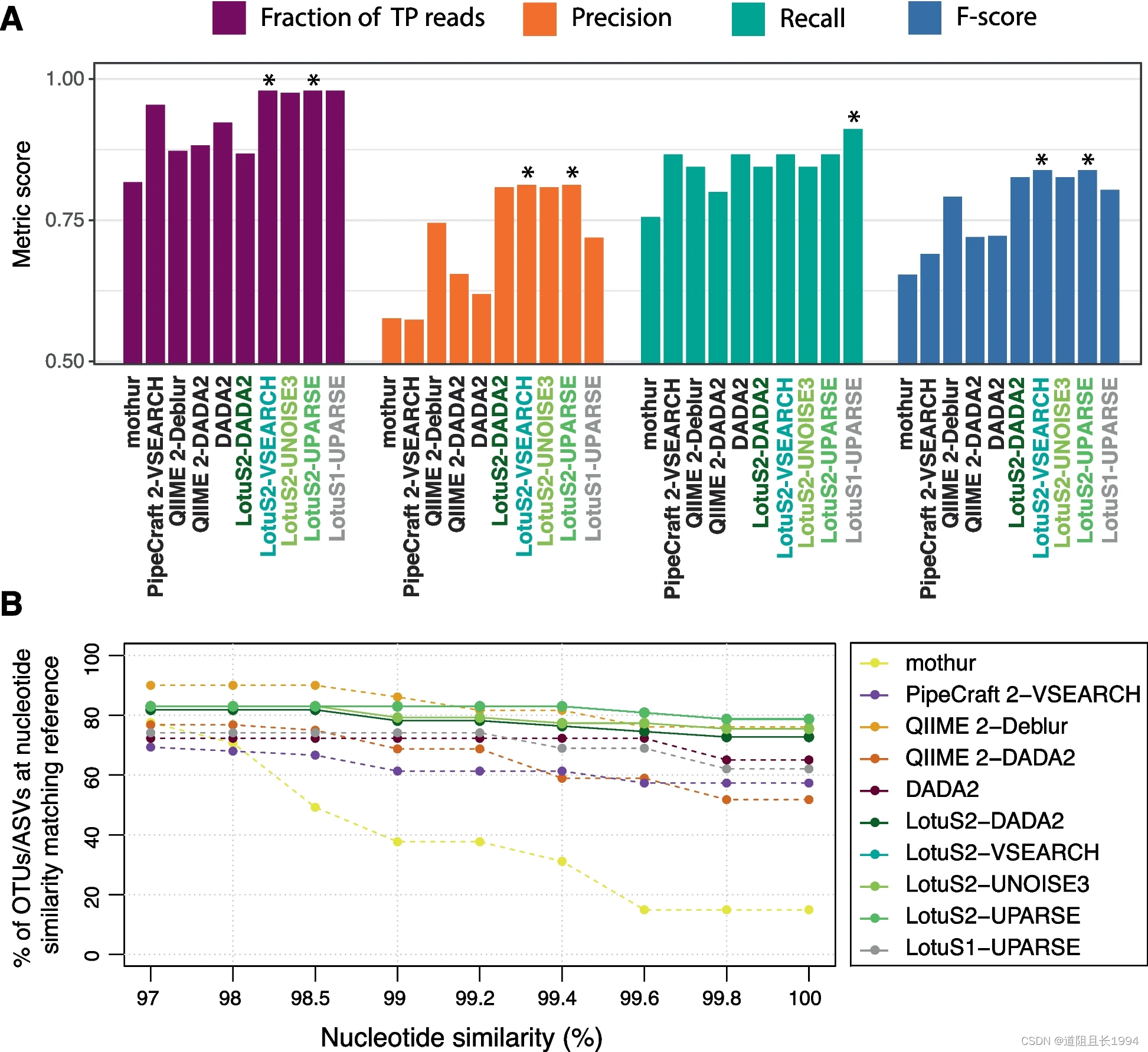
综合以上优点,lotus2完全值得尝试,接下来进入实战部分。
Lotus2的安装
推荐conda安装
conda install -c bioconda lotus2
如果失败了,可以尝试:
conda create -c conda-forge -c bioconda --strict-channel-priority -n lotus2 lotus2
安装成功后激活环境:
conda activate lotus2
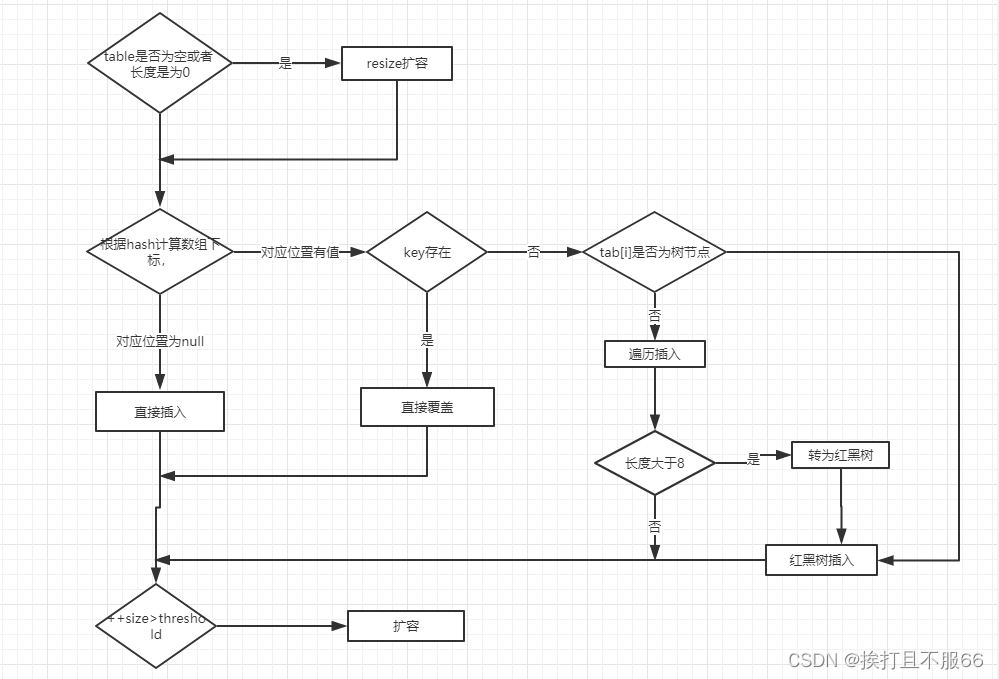
细菌16s扩增子分析运行命令
lotus2 -i Example/ \ #序列所在位置的文件夹
-m Example/miSeqMap.sm.txt \ #map文件,示意文件参考后续内容
-o myTestRun2 \ # 输出文件夹,OTU table、tree、运行日志等都会放在这个文件夹中
-s configs/sdm_miSeq2.txt \ #配置文件,这个是作者提供的,设定了运行命令所需的参数
-p miSeq \ #测序平台,比如我的真菌ITS测序用的是PacBio就填写PacBio即可
-amplicon_type SSU \ #真菌ITS测序则填写ITS
-forwardPrimer GTGYCAGCMGCCGCGGTAA \
-reversePrimer GGACTACNVGGGTWTCTAAT \
-CL dada2 \ #聚类的方法
-refDB SLV \ #注释用的数据库
-taxAligner lambda \ #注释的方法
-t 14 #使用的线程数
map文件的示意,注意测序文件和map文件的所在目录。假如我们所在的工作目录为WD,序列位置为:WD/Example/seq.fastq.gz。
| #SampleID | fastqFile | SequencingRun |
|---|---|---|
| ANG1 | PID-0292-1_S1_L001_R1_001.fastq.sm.gz,PID-0292-1_S1_L001_R2_001.fastq.sm.gz | C |
| ANG2 | PID-0292-1_S2_L001_R1_001.fastq.sm.gz,PID-0292-1_S2_L001_R2_001.fastq.sm.gz | C |
这里C的含义不是很清楚,有了解的小伙伴可以留言一下。
真菌ITS扩增子分析运行命令:
lotus2 -i YOURPATH \
-p PacBio \
-id 0.97 \
-CL cdhit \
-s configs/sdm_PacBio_ITS.txt \
-refDB UNITE \
-m ./map.txt \
-o ./LotuS2 \
-forwardPrimer GTACACACCGCCCGTCG \
-reversePrimer CGCCTSCSCTTANTDATATGC \
-t 14 \
-amplicon_type ITS \
-taxAligner vsearch \
-buildPhylo 0 \ #一般认为ITS构建的遗传发育树不靠谱,可以不用构建。
#Recommendation from author:
#We recommend the cautious usage of the
#phylogenetic tree for ITS because high
#variation of ITS sequences may lead to erroneous trees.